Translate this page into:
Alopecia areata in a girl: A conundrum of endocrinopathies
2 Department of Dermatology, Maulana Azad Medical College, New Delhi, India
Correspondence Address:
Aashima Dabas
Department of Pediatrics, Maulana Azad Medical College, Bahadur Shah Zafar Marg, New Delhi - 110 002
India
| How to cite this article: Tyagi V, Dabas A, Yadav S, Kochhar AM. Alopecia areata in a girl: A conundrum of endocrinopathies. Indian J Dermatol Venereol Leprol 2020;86:295-297 |
Sir,
Alopecia areata is a complex genetic, immune-mediated disease targeting the anagen hair follicles.[1] Autoimmunity seems to be significant etiological factor because of its strong association with other autoimmune disorders like vitiligo, lupus erythematosus, myasthenia gravis, scleroderma, ulcerative colitis, type I diabetes, thyroiditis, celiac disease and rheumatoid arthritis.[1] We report a case of patchy hair loss in an adolescent girl with multiple underlying endocrine abnormalities.
An 11-year-old girl presented to the dermatology department with complaints of patchy hair loss over scalp since 3 years of age which subsequently progressed to alopecia totalis in next 4 years. [Figure - 1]. She had been receiving oral multivitamins and topical preparations for last 8 years without much improvement. We administered oral mini-pulse betamethasone therapy (2–3 mg betamethasone on consecutive days, twice weekly) resulting in some improvement at 11 years of age [Figure - 2]. At 12 years, she presented with weight gain (without goitre) and primary autoimmune hypothyroidism was diagnosed as her serum thyroid-stimulating hormone (TSH) was 7.05 IU/mL, free thyroxine (fT4) was 0.95 ng/mL, free T3 was 2.88 pg/mL and antithyroid peroxidise was> 1300 IU/mL. Oral thyroxine was supplemented to her weekly steroid pulses at 50 μg daily. Her growth velocity remained 3 cm/year despite normalization of thyroid function at 13 years of age. The child presented with delayed puberty and poor height gain after 1 year. Anthropometry showed a height of 142.1 cm (standard deviation score (SDS) -1.98), weight 44 kg (SDS -0.33), and skeletal age of 10 years. All routine investigations were unremarkable. Growth hormone stimulation tests showed a peak growth hormone level of 7.1 ng/mL suggestive of deficiency. Gonadotropin-releasing hormone stimulation test revealed reduced serum follicle-stimulating hormone, luteinizing hormone, and estradiol levels (3.1 mIU/mL, 0.5 mIU/mL, and <5.0 pg/mL, respectively) indicating hypogonadotropic hypogonadism. Serum cortisol (after ACTH stimulation test) and repeat thyroid function tests were normal. Magnetic resonance imaging of the brain showed small-sized pituitary gland (2 mm) suggestive of hypoplasia secondary to hypophysitis [Figure - 3]. She was started on oral estradiol valerate for pubertal induction which led to catch-up in height. The first-degree relatives were not screened due to lack of significant family history. She was continued on thyroxine and weekly steroid pulses. There were no ocular side effects or evidence of impaired glucose metabolism in the index case.
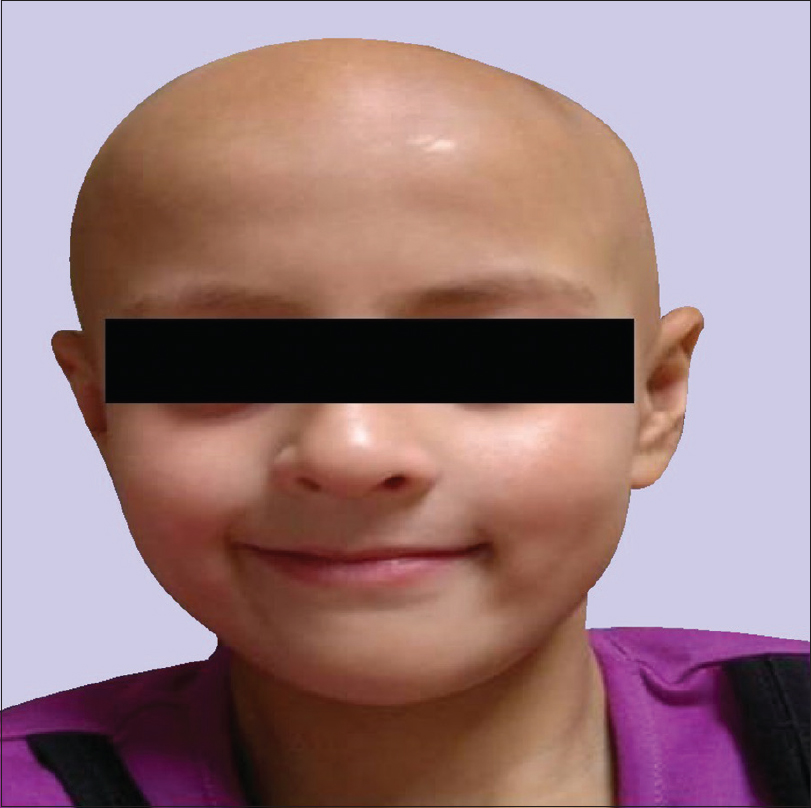 |
| Figure 1: Alopecia totalis at 7 years of age |
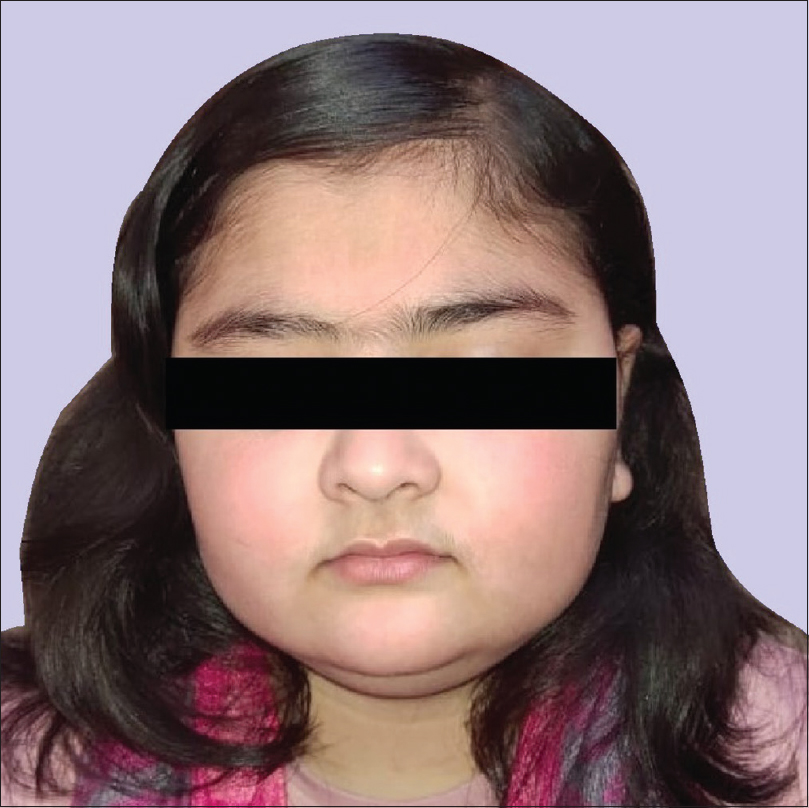 |
| Figure 2: Good hair growth at 11 years of age |
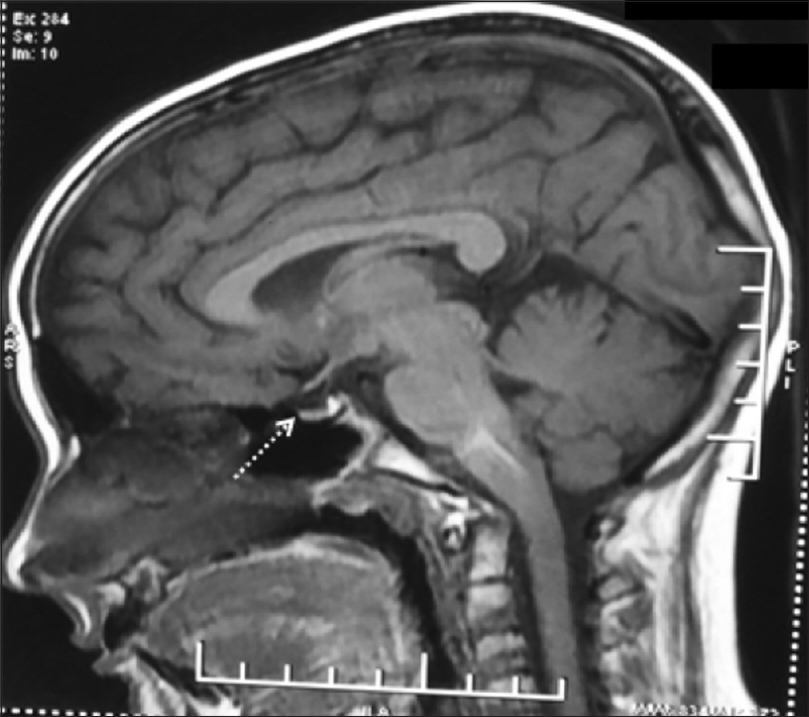 |
| Figure 3: Magnetic resonance imaging brain (sagittal section) showing hypoplastic pituitary |
Several endocrine diseases can manifest in patients with alopecia areata, thyroid diseases (Hashimoto's thyroiditis, Graves' disease and goitre) being the commonest, reported in 2% to 28% cases.[4],[5] Other rare endocrine abnormalities include growth hormone deficiency[6] and hypogonadotropic hypogonadism[7],[8],[9] with autoimmunity being the possible etiological cause.[7],[9] The index case had multiple anterior pituitary hormone deficiency along with alopecia areata, the possible insult being autoimmune hypophysitis. The clinical picture of hypophysitis was supported by the radiological finding of hypoplastic pituitary on MRI. Ajith et al. and Bansal et al. reported similar cases of panhypophypopituitarism with alopecia areata in older individuals.[8],[9]
The coexistence of multiple autoimmune disorders suggests a spectrum of polyglandular autoimmune syndrome. These syndromes show great heterogeneity and manifest sequentially with a large time interval between the occurrence of the first and second glandular autoimmune diseases. Polyglandular autoimmune syndrome also clusters with several nonendocrine autoimmune diseases. It has been classified into juvenile and adult type, the later being subclassified into four types. polyglandular autoimmune syndrome type I may initially present as chronic mucuocutaneous candidiasis followed by hypoparathyroidism and adrenal insufficiency along with other nonendocrine autoimmune diseases. The major endocrine component of polyglandular autoimmune syndrome II is Addison disease, while type III usually includes type 1 diabetes and autoimmune thyroid disease. polyglandular autoimmune syndrome type IV is extremely heterogeneous involving a large variety of glandular autoimmune diseases not included within polyglandular autoimmune syndrome II– III.[10] The index patient might satisfy the criteria of polyglandular autoimmune syndrome type IV; however, this entity is rare in children.
The rarity of the case lies not only in the varied endocrinopathies involved but also in the age of presentation. This case highlights the need of close monitoring of growth and pubertal development in dermatological conditions especially alopecia to detect endocrinal complications and manage them appropriately. Screening for polyglandular autoimmunity is also relevant in patients with monoglandular autoimmune disease and/or first-degree relatives of patients with polyglandular autoimmune syndrome.[10]
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form, the legal guardian has given her consent for images and other clinical information to be reported in the journal. The guardian understands that names and initials will not be published and due efforts will be made to conceal identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
| 1. |
Hordinsky MK. Overview of alopecia areata. J Investig Dermatol Symp Proc 2013;16:S13-5.
[Google Scholar]
|
| 2. |
Goh C, Finkel M, Christos PJ, Sinha AA. Profile of 513 patients with alopecia areata: Associations of disease subtypes with atopy, autoimmune disease and positive family history. J Eur Acad Dermatol Venereol 2006;20:1055-60.
[Google Scholar]
|
| 3. |
Patel D, Li P, Bauer AJ, Castelo-Soccio L. Screening guidelines for thyroid function in children with alopecia areata. JAMA Dermatol 2017;153:1307-10.
[Google Scholar]
|
| 4. |
Tan E, Tay YK, Goh CL, Chin Giam Y. The pattern and profile of alopecia areata in Singapore – A study of 219 Asians. Int J Dermatol 2002;41:748-53.
[Google Scholar]
|
| 5. |
Cunliffe WJ, Hall R, Stevenson CJ, Weightman D. Alopecia areata, thyroid disease and autoimmunity. Br J Dermatol 1969;81:877-81.
[Google Scholar]
|
| 6. |
Yasutomo Y, Yoshida A, Noritake M, Nemoto Y, Kugai N, Nagata N. Simultaneous occurrence of SIADH, secondary hypogonadism and alopecia universalis in a woman with IDDM. Endocrinol Jpn 1991;38:445-9.
[Google Scholar]
|
| 7. |
Slti IS, Salem Z. Familial hypogonadotropic hypogonadism with alopecia. Can Med Assoc J 1979;121:428-30, 433-4.
[Google Scholar]
|
| 8. |
Ajith C, Gupta S, Bhansali A, Radotra BD, Kanwar AJ, Kumar B, et al. Alopecia areata associated with idiopathic primary hypophysitis. Clin Exp Dermatol 2005;30:250-2.
[Google Scholar]
|
| 9. |
Bansal S, Kumar N, Kochhar AM, Ghai R. Alopecia universalis in a patient with Sheehan's syndrome. Indian Dermatol Online J 2016;7:58-9.
[Google Scholar]
|
| 10. |
Kahaly GJ, Frommer L. Polyglandular autoimmune syndromes. J Endocrinol Invest 2018;41:91-8.
[Google Scholar]
|
Fulltext Views
2,776
PDF downloads
1,185





![[Figure - 1]](#fig_ijdvl_2020_86_3_295_278566_f1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2020_86_3_295_278566_f2.jpg){kind=link}
![[Figure - 3]](#fig_ijdvl_2020_86_3_295_278566_f3.jpg){kind=link}