Translate this page into:
Ankyloblepharon–ectodermal dysplasia–clefting syndrome
Correspondence Address:
Surendra Kumar
Quarter Number - AC III/3, JLN Hospital Campus, Near Savitri Girls College, Ajmer - 305 001, Rajasthan
India
| How to cite this article: Ramya B G, Gondane S, Kumar S, Dangi S. Ankyloblepharon–ectodermal dysplasia–clefting syndrome. Indian J Dermatol Venereol Leprol 2015;81:629-630 |
Sir,
A 3 week old boy born by full-term normal vaginal delivery presented with fever and urinary retention of 2 days duration. He was also suffering from multiple abnormalities of the face and eyelids since birth. The boy was reported to have multiple erosions all over the body, more pronounced on the scalp. The erosions healed following the application of fusidic acid 2% cream. At presentation, he only had erosions on the frontal and temporal scalp. There was no family history of consanguinity and the prenatal course was uneventful. Physical examination revealed erosions on the scalp and trunk with scanty hair. Eyebrows and eyelashes were absent and the upper and lower eyelids were fused together (ankyloblepharon) [Figure - 1]. He also had a broad nose, dystrophic nails [Figure - 2], cleft lip, cleft palate [Figure - 3], and hypospadias [Figure - 2].
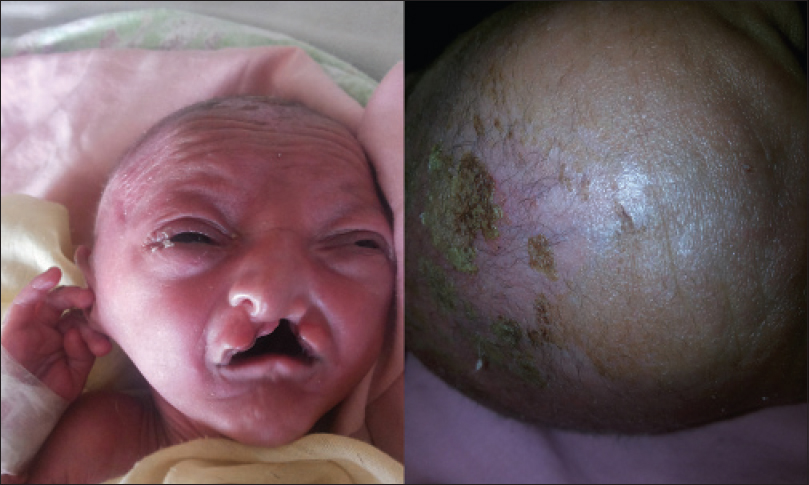 |
| Figure 1: Ankyloblepharon and scalp erosions |
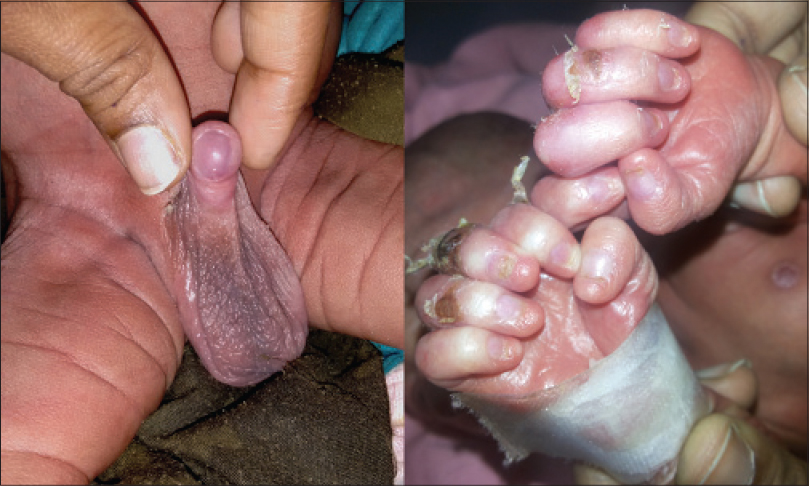 |
| Figure 2: Hypospadias and nail dysplasia |
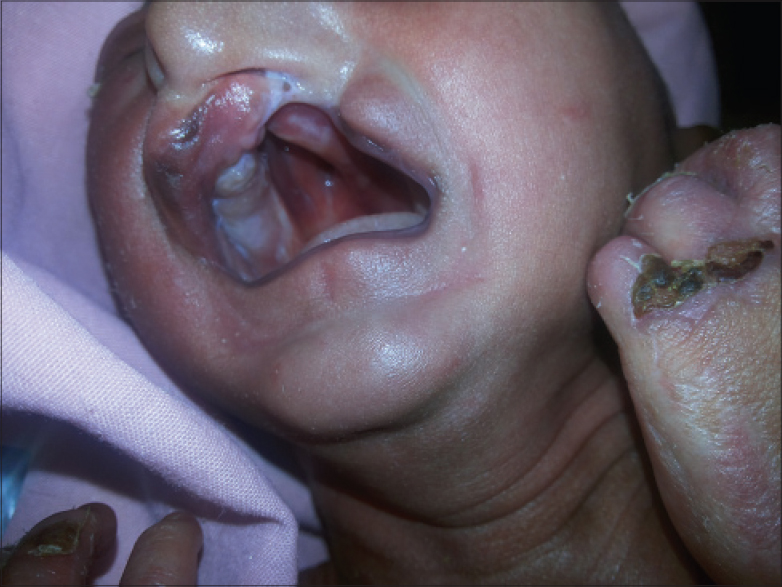 |
| Figure 3: Clefting of the upper lip and palate |
Routine investigations were within normal limits, other than elevated white blood cell counts. Blood culture grew coagulase-negative staphylococcus indicating a hospital acquired infection. Ultrasonography showed dilatation of the pelvi-calyceal system and a distended bladder, which was probably secondary to hypospadias. Based on the clinical features, we made the diagnosis of ankyloblepharon-ectodermal defects-cleft lip/palate (AEC) syndrome, a type of ectodermal dysplasia.
Ectodermal dysplasias refer to a group of diseases in which there is a defect in the development of the hair, teeth, nails, sweat glands or other structures originating from the ectoderm. Ankyloblepharon-ectodermal defects-cleft lip/palate syndrome (also known as Hay-Wells syndrome, after the two physicians who described it in 1976), is a rare autosomal dominant genetic disorder with unknown prevalence characterized by ankyloblepharon filiforme adnatum (partial thickness fusion of the eyelid margins), cleft lip/palate, severe scalp erosions and abnormalities of the epidermal appendages including hypotrichosis, hypodontia, absent or dystrophic nails and mild hypohidrosis.[1],[2] Other findings include supernumerary nipples and ectopic breast tissue, malformed auricles, recurrent otitis media, secondary conductive deafness and palmoplantar hyperkeratosis.[3],[4]
The syndrome is inherited in an autosomal dominant manner. Approximately 30% of individuals have an affected parent and 70% have a sporadic mutation.[1] Cases with autosomal recessive inheritance and germline mosaicism are also reported.[5] In our case, the mutation was most likely sporadic as all the other family members were healthy.
Ankyloblepharon-ectodermal defects cleft lip/palate syndrome is most often a clinical diagnosis although in some instances molecular genetic testing of the causative gene, TP63 can be helpful in establishing the diagnosis. This syndrome should be differentiated from other ectodermal dysplasias such as ectrodactyly-ectodermal dysplasia-cleft lip/palate syndrome which is characterized by bony hand and foot abnormalities and lacks ankyloblepharon, curly hair and nail dysplasia. Rapp-Hodgkin syndrome, with many clinical similarities was once thought to be a distinct entity; it is now considered to be allelic to ankyloblepharon-ectodermal defects cleft lip/palate syndrome. Erosions on the scalp with formation of granulation tissue and the absence of ankyloblepharon distinguish Rapp–Hodgkin syndrome from ankyloblepharon-ectodermal defects cleft lip/palate syndrome. The presence of erythroderma and extensive erosions in the postnatal period can lead to the misdiagnosis of epidermolysis bullosa.[1],[3] We were able to find two previous reports of ankyloblepharon-ectodermal defects cleft lip/palate syndrome from India.[2],[4]
Therapy should be multidisciplinary and focused on early lysis of ankyloblepharon and surgical correction of cleft lip/palate at the appropriate age. Management of infections and skin care with light emollients, appropriate dressings and gentle removal of scales and crusts is essential. The importance of early diagnosis of this syndrome should be emphasized in order to implement appropriate genetic counseling for parents as well as for the timely treatment of the patient.[1]
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
| 1. |
Sutton VR, Bree AF, Bokhoven H. Ankyloblepharon-ectodermal defects-cleft lip/palate syndrome. In: Pagon RA, Adam MP, Ardinger HH, editors. GeneReviews®. Seattle (WA): University of Washington, Seattle; 1993-2015. Available from: http://www.genetests.org. [Last accessed on 2014 Dec 15].
[Google Scholar]
|
| 2. |
Nagaveni NB, Umashankara KV. Hay-Wells syndrome of ectodermal dysplasia: A rare autosomal dominant disorder. Indian J Hum Genet 2011;17:245-6.
[Google Scholar]
|
| 3. |
Bree AF, Agim N, Sybert VP. Ectodermal dysplasias. In: Goldsmith LA, Katz SI, Gilchrest BA, Paller AS, Leffell DJ, Wolff K, editors. Fitzpatrick's Dermatology in General Medicine. 8th ed. New York: McGraw-Hill; 2012. p. 1691-703.
[Google Scholar]
|
| 4. |
Kulkarni ML, Deshmukh S, Matani D, Gayatri K. Hay-wells syndrome of ectodermal dysplasia. Indian J Pediatr 2006;73:101.
[Google Scholar]
|
| 5. |
Barbaro V, Nardiello P, Castaldo G, Willoughby CE, Ferrari S, Ponzin D, et al. A novel de novo missense mutation in TP63 underlying germline mosaicism in AEC syndrome: Implications for recurrence risk and prenatal diagnosis. Am J Med Genet A 2012;158A: 1957-61.
[Google Scholar]
|
Fulltext Views
3,590
PDF downloads
961





![[Figure - 1]](#fig_ijdvl_2015_81_6_629_168333_f1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2015_81_6_629_168333_f2.jpg){kind=link}
![[Figure - 3]](#fig_ijdvl_2015_81_6_629_168333_f3.jpg){kind=link}