Translate this page into:
Cerebrotendinous xanthomatosis: Need for early diagnosis
Correspondence Address:
K Muhammed
Kunnummal House, Koroth School Road, Vatakara - 673101, Kozhikode, Kerala
India
| How to cite this article: Muhammed K, Nandakumar G, Saritha S. Cerebrotendinous xanthomatosis: Need for early diagnosis. Indian J Dermatol Venereol Leprol 2006;72:364-366 |
Abstract
Cerebrotendinous xanthomatosis is a rare autosomal recessive lipid storage disease characterized by widespread tissue deposition of two neutral sterols, cholestanol and cholesterol, resulting in tendinous xanthomas, juvenile cataracts, progressive neurological defects and premature death from arteriosclerosis. The primary biochemical defect is deficiency of hepatic mitochondrial enzyme sterol-27-hydroxylase which catalyses the hydroxylation of cholestanol (5-alpha dehydro derivative of cholesterol) and this deficiency decreases bile acid synthesis. Substantial elevation of serum cholestanol and urinary bile alcohols with low to normal plasma cholesterol concentration establishes the diagnosis. Cerebrotendinous xanthomatosis is exceptionally rare in the Indian population. We are reporting a woman with this rare disorder, who was on antiepileptic and antipsychotic drugs for a prolonged period and whose original condition went undiagnosed. She presented with xanthomas on the Achilles tendons and the upper end of tibia. She was mentally subnormal and her serum cholestanol level was raised. Her younger sister too was severely affected by this disorder. Early treatment with chenodeoxycholic acid is known to prevent disease progression.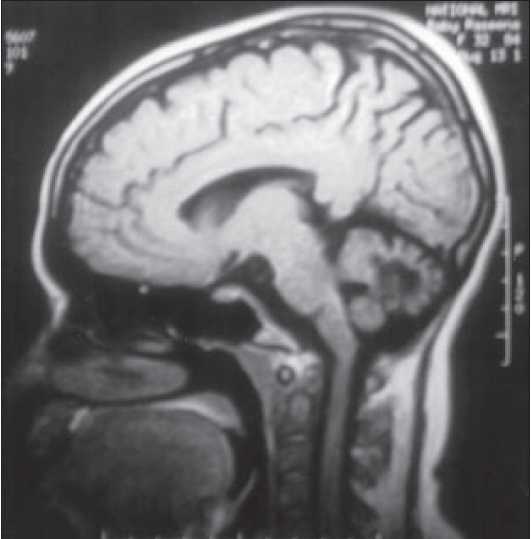 |
| MRI scan showing severe cerebellar atrophy |
|
| MRI scan showing severe cerebellar atrophy |
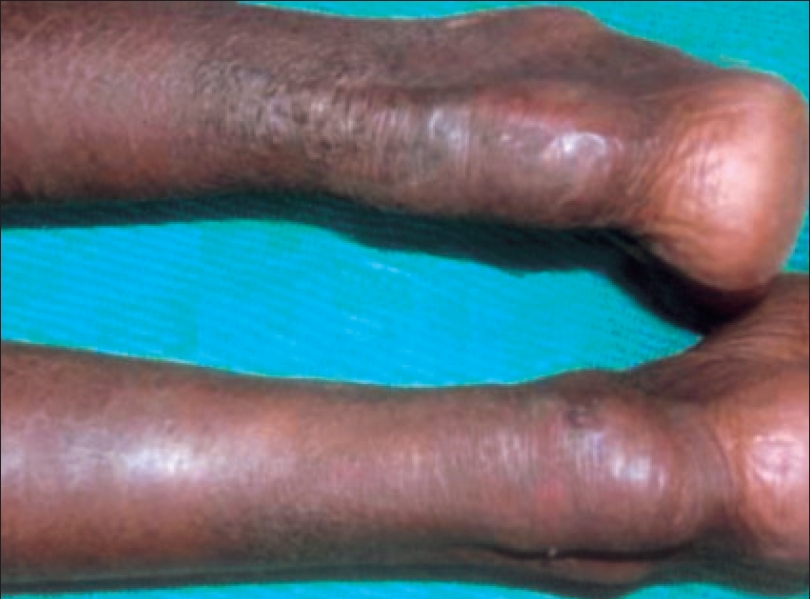 |
| Tendinous xanthomas on both Achilles tendons |
|
| Tendinous xanthomas on both Achilles tendons |
Introduction
Cerebrotendinous xanthomatosis (CTX) or cholestanol lipidosis is a rare autosomal recessive metabolic disease.[1],[2] A review of the literature revealed 175 patients with documented CTX of which 56% were females.[3] Several genetic studies have shown mutations in the sterol 27-hydroxylase gene (CYP 27 gene) resulting in markedly diminished activity of the enzyme sterol 27-hydroxylase in CTX patients.[3],[4] Sterol 27-hydroxylase enzyme catalyses the hydroxylation of cholestanol and its deficiency leads to elevated plasma cholestanol levels and consequently its accumulation in the brain, lens, tendons and other tissues.[1],[3],[4]
Diagnosis is confirmed by elevated plasma and bile cholestanol[1] and increased levels of urinary bile alcohols.[5] Treatment of choice is chenodeoxycholic acid.[1],[4] Cerebrotendinous xanthomatosis is exceptionally rare in the Indian population. Most cases have been reported in journals on neurology or metabolism with only four cases reported in dermatology literature.[5] We are reporting a case of CTX in a family of consanguineous parentage, with severer involvement of the younger sister.
Case report
A 26-year-old mentally retarded woman attended our outpatient department with complaints of swellings along the course of both Achilles tendons of 16 years duration [Figure - 1] and at the upper end of left tibia of three years duration. They were asymptomatic and gradually increasing in size. The patient gave history of surgery for bilateral mature cataract at the age of eight years. She developed hyperirritable behavior and poor scholastic performance so that she stopped going to school from fourth standard. She had gait ataxia with positive Rhomberg′s sign. She had generalized xerosis of the skin and asteatotic eczema of legs.
The patient was the eldest of four siblings of consanguineous parentage. Her 24-year-old younger sister had history of seizures since the first month of life and had bilateral mature cataract from an early age. She also had a gradual decline of mental function, being bedridden at the present and seemed more severely affected than the index case. She had developed Achilles tendon swellings three years ago. Her 20-year-old brother was on phenytoin sodium for grand mal seizures for the past three years while another 18-year-old brother was clinically unaffected.
The Achilles tendon swellings of both women and the swelling on the left tibial tuberosity of the index case were proved by biopsy to be tendinous xanthomas. Magnetic resonance imaging (MRI) of the index patient′s brain revealed severe cerebellar atrophy [Figure - 2], abnormal signals in the dentate nuclei and cerebellar white matter (hyperintense lesion in the T2 weighted images) with mild cerebral atrophy. A peripheral rim of marked hypointensity was seen around the hyperintense areas caused by xanthomas. Serum cholestanol of the patient (by partition chromatography and spectral photometric analysis) was elevated (3.8 ng/dl) which confirmed the diagnosis of cerebrotendinous xanthomatosis. Serum cholestanol of her epileptic brother was within normal limits (normal < 1 ng/dl).
Discussion
Cerebrotendinous xanthomatosis is a rare familial sterol storage disease.[1],[3] The primary biochemical defect is deficiency of hepatic mitochondrial enzyme sterol-27-hydroxylase which catalyses the hydroxylation of cholestanol (5-alpha dehydro derivative of cholesterol) and its deficiency decreases bile acid synthesis. This reduces feedback inhibition on cholesterol 7-alpha hydroxylase, which is the rate-limiting enzyme, resulting in synthesis and accumulation of more cholestanol.[4] Of the 175 documented cases of CTX, 71% had tendon xanthomas, 81% had low intelligence and incidence of cataracts and other neurological symptoms were seen in 92% and 100% cases respectively.[3] Patients usually present in childhood or early adult life.[1]
Tendon xanthomas, especially over the Achilles tendon are characteristic of the disorder and clinically resemble those seen in familial hypercholesterolemia or hyperlipoproteinemia but biochemical analysis reveals that they contain high amounts of cholestanol and little cholesterol. Mental retardation and progressive spasticity may develop.[1],[2]
Other reported manifestations of this rare syndrome include juvenile cataract,[1],[4],[6] abnormal behavior [2] and premature arteriovascular disease.[1],[7] In our case two of the four siblings had features of CTX. Both had tendon xanthomas, juvenile cataract, behavior abnormalities and progressive neurological problems mainly of cerebellar origin. As in our case, psychiatric manifestations may occur in patients with CTX.[2] These include depressed mood, irritability, poor appetite, insomnia, fatigability and pessimistic thinking.[2] These manifestations should be treated with antipsychotic drugs along with specific treatment.[2]
The diagnosis of CTX can be made biochemically by detecting increased serum levels of cholestanol[1] or urine bile alcohol[5] and genetically by detecting molecular defects in sterol-27-hydroxylase gene.[5] Conventional MRI studies have shown focal/ diffuse white matter abnormalities and different degrees of cerebral and cerebellar atrophy in the brain of patients with CTX. The bilateral nonhomogenous, hyperintense magnetic resonance signal in dentate nuclei and surrounding cerebellar white matter, can be considered as a neuroradiological feature suggestive of CTX and could become an important diagnostic marker.[8] The demyelinated areas appear hyperintense and the peripheral rim of marked hypointensity is caused by the xanthoma.[8]
CTX is very rare in the Indian population. In1999, Gobinda et al . reported CTX in two siblings from an Indian family and Gaikwad et al . described neuroimaging findings in two siblings from India.[8]
Several modes of treatment have been tried for CTX. Since 1975, chenodeoxy cholic acid (CDCA) 750 mg daily has been commonly used as the standard therapy, which influences the negative feedback of cholesterol and bile acid synthesis. There is a considerable decrease in the serum cholestanol and a sharp decline in the excretion of urine bile alcohols.[5] But a combination of CDCA with 3-hydroxy 3-methyl glutaryl coenzyme A (HMG COA) reductase inhibitors such as pravastatin or simvastatin (10-40 mg daily) is found to be more effective in lowering the serum cholestanol levels.[1] Long-term treatment may arrest or even reverse the progression of the disease.
Unfortunately, as in our case, the disease is not usually diagnosed before the second or third decade of life and at this stage cholestanol has already been extensively deposited in many tissues. Therefore, early diagnosis of this rare metabolic disease is necessary and CTX should be considered in every patient with intellectual impairment, spastic - ataxic signs, juvenile cataract and tendon xanthomas and MRI imaging should be done as soon as possible.
| 1. |
Black MM, Gawkrodger DJ, Seymour CA, Weismann K. Metabolic and nutritional disorders. In : Rook, Wilkinson, Ebling, editors. Text book of Dermatology. 6th ed. Blackwell Science: London; 1998. p. 2577-677.
[Google Scholar]
|
| 2. |
Lee Y, Lin PY, Chin NM, Chang WN, Wen JK. Cerebrotendinous Xanthomatosis with psychiatric disorders: Report of three siblings and literature review. Chang Gung Med J 2002;25: 334-40.
[Google Scholar]
|
| 3. |
Moghadasian MH. Cerebrotendinous xanthomatosis: Clinical course, genotype and metabolic backgrounds. Clin Invest Med 2004;27:42-50.
[Google Scholar]
|
| 4. |
Seyama Y. Cholesterol metabolism, molecular pathology and nutritional implications. J Med Food 2003;6:217-24.
[Google Scholar]
|
| 5. |
Bel S, Garcia-Patos V, Rodriguez L, Selvan A, Diaz P, Wolthers BG, et al . Cerebrotendinous xanthomatosis. J Am Acad Dermatol 2001;45:292-5.
[Google Scholar]
|
| 6. |
Campdelacreu J, Munoz E, Cervera A, Jauma S, Giros M, Tolosa E. Cerebrotendinous xanthomatosis without tendinous xanthomas: Presentation of two cases. Neurologia 2002;17: 647-50.
[Google Scholar]
|
| 7. |
Valdivielso P, Calandra S, Durcan JC, Garuti R, Herrera E, Gonzalez P. Coronary heart disease in a patient with cerebrotendinous xanthomatosis. J Intern Med 2004:255:680-3.
[Google Scholar]
|
| 8. |
Gaikwad SB, Garg A, Mishra NK, Gupta V, Srivastava A, Sarkar C. Cerebrotendinous xanthomatosis: Neuroimaging findings in two siblings from an Indian family. Neurol India 2003;51:401-3.
[Google Scholar]
|
Fulltext Views
2,961
PDF downloads
1,749





![[Figure - 1]](#fig_ijdvl_2006_72_5_364_27754_1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2006_72_5_364_27754_2.jpg){kind=link}