Translate this page into:
Costello syndrome
Correspondence Address:
J Madhukara
Department of Dermatology, St John's Medical College Hospital, Sarjapur Road, Bangalore - 560 034
India
| How to cite this article: Madhukara J, Kumaran M S. Costello syndrome. Indian J Dermatol Venereol Leprol 2007;73:406-408 |
Abstract
Costello syndrome is a rare, distinctive, multiple congenital anomaly syndrome, characterized by soft, loose skin with deep palmar and plantar creases, loose joints, distinctive coarse facial features and skeletal and cardiac abnormalities. The affected patients have a predisposition to develop malignancy, developmental delays and mental retardation. Recently, a 7-year-old male child born to normal nonconsanguineous parents presented to us with abnormal facial features, arrhythmia, mitral valve dysfunction and growth retardation. His cutaneous examination revealed lax and pigmented skin over hands and feet with deep creases, acanthosis nigricans and short curly hairs. Its differentiation from other syndromes with similar clinical features is discussed in this article.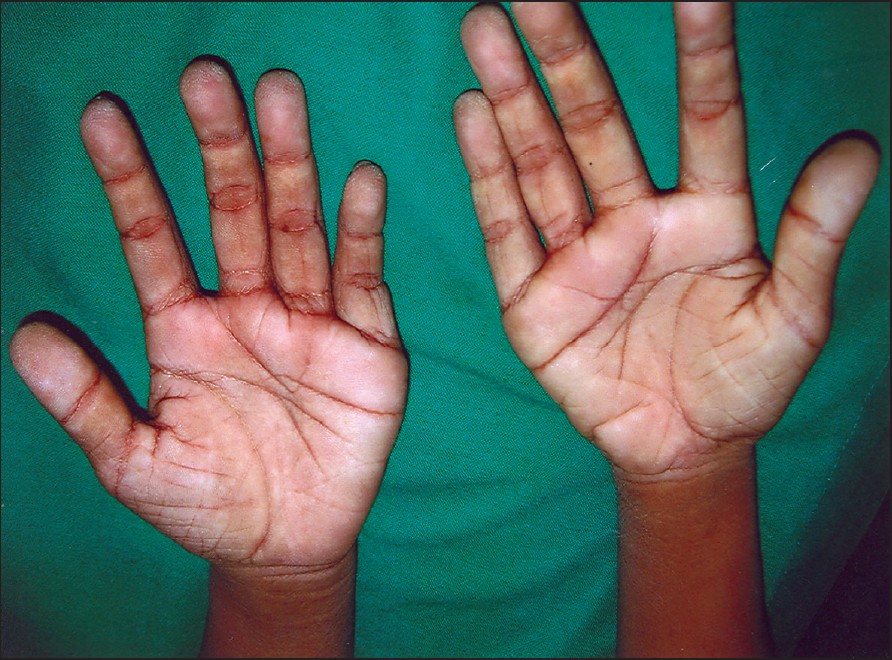 |
| Figure 4: Hyperkeratosis of palms and soles |
|
| Figure 4: Hyperkeratosis of palms and soles |
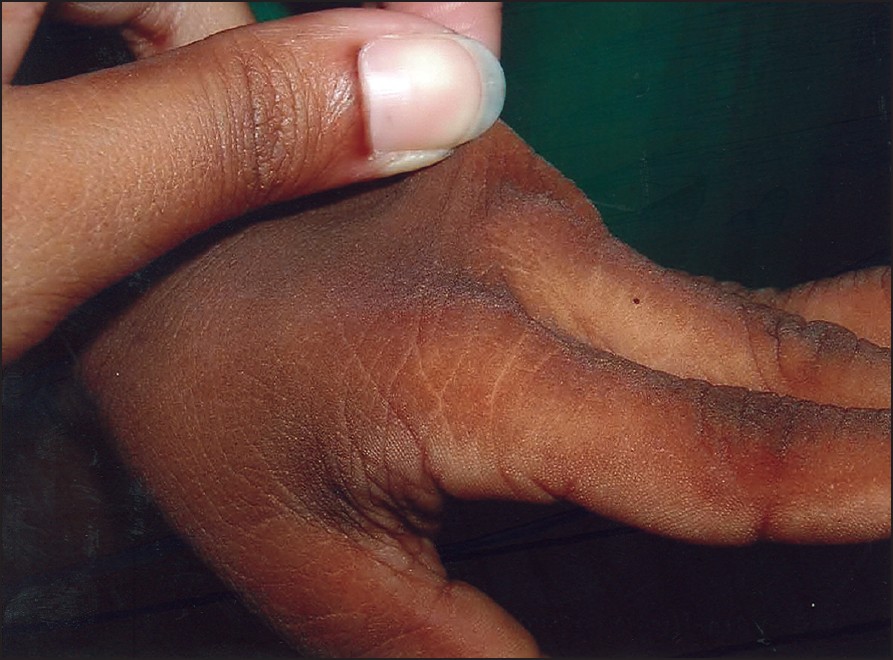 |
| Figure 3: Acanthosis of the skin with laxity |
|
| Figure 3: Acanthosis of the skin with laxity |
 |
| Figure 2: Dysmorphic facies in Costello syndrome |
|
| Figure 2: Dysmorphic facies in Costello syndrome |
 |
| Figure 1: Sparse curly hair in Costello syndrome |
|
| Figure 1: Sparse curly hair in Costello syndrome |
Introduction
Costello first described Costello syndrome (CS) in 1977, while observing two unrelated children with deficient growth, loose skin, coarse facial features and nasal papillomata. [1] The condition, though rare, is well characterized. It often presents initially to a cardiologist; however, knowledge about the clinical features of this condition may help dermatologists in early recognition and management of potentially serious complications. We discuss a case of CS recently seen by us.
Case Report
A 7-year-old male child born to normal nonconsanguineous parents was referred from a cardiac referral hospital with a diagnosis of Costello/Noonan syndrome for cutaneous evaluation. Mother had an uneventful gestation and a normal labor. After birth, the boy was noted to have abnormal facial features. A few days later, he had severe respiratory distress and cyanosis. On evaluation at a local hospital, he was found to have arrhythmias and mitral valve dysfunction for which treatment was started. His mother reported of weak suckling reflex, poor weight gain following which there was growth retardation and short stature. He was the only sibling with no family history of similar complaints.
Clinical examination revealed a short-statured boy with a low IQ and partial valgus deformity of both hands and legs. The boy had relative macrocephaly with thin, curly and sparse hair [Figure - 1]. His face had coarse features with low-set ears, mild ptosis and a minimally depressed nasal bridge with wide nostrils, thick lips and low frontal hairline [Figure - 2]. The skin was lax more so over hands and feet, pigmented and rough. The skin over the dorsa of hand and fe et al so showed thickening of skin along with short, hyper-extensible fingers and toes with brittle nails. Acanthosis nigricans was seen around axillae, neck and dorsa of hands [Figure - 3]. The palms and soles were hyperkeratotic with deep creases [Figure - 4]. Eye and ear examinations and routine biochemical investigations were within normal limits. Skin biopsy was not done, as patient′s attenders were unwilling. The cutaneous evaluation was suggestive of CS.
Discussion
Costello syndrome is characterized by failure to thrive, relative macrocephaly and characteristic facies. Infants are usually born at term and have normal birth length and weight, but this is followed by postnatal growth failure resulting in short stature. [2],[3] They have a distinctive coarse face with relative macrocephaly, fine, sparse, curly, kinky hair, palpebral ptosis, downward-slanting palpebral fissures, epicanthal folds, strabismus, fleshy thickened lobes, posterior rotation of the pinnae and low-set ears with prominent helices. Affected patients have short nose with broad nasal base, macroglossia, thick lips and high arched palate. [1],[4],[5] These patients usually have mild to moderate intellectual disability, but some can develop severe intellectual impairment. Their IQ at 10 years of age often equals 40-49. [2]
Loose redundant skin of neck, dorsa of hands and feet is noticeable at birth. [1],[4] Patients with the CS also show palmoplantar hyperkeratosis with deep creases. [1] Palmoplantar hyperkeratosis spares pressure zones. [4] Skin tags, acanthosis nigricans and nail dystrophy with koilonychia are seen as the most common skin and adnexal anomalies. Most of these cutaneous findings were also seen in our patient except for the papillomas. Papillomas are soft fibrous out growths that are usually seen over perinasal and perioral areas. [5] They have also been reported around perianal areas. [1] Although papillomas are characteristic findings, they can appear anytime in life. Hence in our patient, the papillomatous growth could appear at a later age. [1],[2]
Atrial tachycardia or other arrhythmias occur in> 30% of patients with the CS, particularly in infancy. Hypertrophic cardiomyopathy and thickened mitral valve are also seen in CS. Tight Achilles tendon and hyperextensible fingers are some of the cardinal manifestations along with generalized joint hypermobility and congenital club feet. [1],[5]
Etiopathogenesis has been elusive; previous studies with cultured fibroblasts from individuals with CS demonstrate excessive accumulation of chondroitin sulfate-bearing proteoglycans, associated with both impaired formation of elastic fibers and an unusually high rate of cellular proliferation. Although elastin deposition is impaired in the skin of these individuals, the defect occurs at a step post transcription. Elastogenesis in the cells of patients with CS is found to be impaired because of a deficiency of elastin-binding protein (EBP), a chaperone that brings tropoelastin to its site of extracellular assembly. [6]
The discovery of mutations in the HRAS gene on chromosome 11p13.3 has decreased the enigma of CS. In most cases, heterozygous mis-sense mutations in the proto-oncogene HRAS are the cause. [7],[8] Although most cases are sporadic, an autosomal dominant mode of inheritance is suggested. [9]
Skin biopsy usually demonstrated normal elastic tissue, although there are reports of abnormal elastic fibers in biopsy. [2] Decrease in collagen with normal elastin is also described. Papillomas have changes typical of warts. [1],[5]
Costello syndrome has to be differentiated from Noonan and cardio-facio-cutaneous (CFC) syndrome; all three syndromes have clinically similar features and are sometimes very difficult to separate one from the other and in such cases only gene mapping helps. Noonan syndrome can be differentiated by presence of dolichocephaly, tall forehead, small chin, hypertelorism and blue-green iris, absence of ear lobe creases, deep philtrum and PTPN11mutation. CFC syndrome can be differentiated by presence of tall forehead with narrowing temples, deep philtrum, ulerythema ophryogenes, palmoplantar keratoderma over pressure sites and lymphedema. [4] Apart from these, cutis laxa should also be considered in differential diagnosis.
Systemically, CFC and Noonan′s differ from CS by presence of severe mental retardation (CFC), learning difficulty (NS ), pulmonary artery stenosis, absence of atrial tachyarrhythmia and presence of transient thrombocytopenia. [1],[2],[4],[5] Prenatal diagnosis by 2D and 3D ultrasonography can show overlap of features of severe Noonan syndrome and CS. [10] Patients with CS have an increased risk for malignant tumors, [6],[9],[10] and risk is suggested to be about 17%. [7] The most common tumor in CS is rhabdomyosarcoma (RMS), followed by neuroblastoma and bladder carcinoma. Benign tumors including multiple ductal papillomata, fourth ventricle mass thought to be a choroid plexus papilloma have also been reported. [3] Endocrine problems like osteoporosis, central hypogonadism and delayed puberty are also described.
The increased tumor frequency in CS led to the proposal of a screening protocol, consisting of abdominal and pelvic ultrasounds, urine studies for catecholamine metabolites and hematuria. [3] Life span is unknown, but presence of CVS anomalies and development of tumors are associated with poor outcome. [2]
Although often described in genetic literature, the cases are scarcely reported in dermatology journals. To the best of our knowledge, this is the probably the first report of CS from India[11].
| 1. |
Torrelo A, L�pez-Avila A, Mediero IG, Zambrano A. Costello syndrome. J Am Acad Dermatol 1995;32:904-7.
[Google Scholar]
|
| 2. |
Toriello HV. Premature aging syndromes. In : Harper J, Orange A, Prose N, editors. Text book of Peadiatric Dermatology. 2nd ed. Blackwell Publishing Limited; 2006. p. 1552-3.
[Google Scholar]
|
| 3. |
Gripp KW. Tumor predisposition in Costello syndrome. Am J Med Genet C Semin Med Genet 2005;137:72-7.
[Google Scholar]
|
| 4. |
Roberts A, Allanson J, Jadico SK, Kavamura MI, Noonan J, Opitz JM, et al . The cardio facio cutaneous syndrome. J Med Genet 2006;43:833-42.
[Google Scholar]
|
| 5. |
Sybert VP. Costello syndrome. In : Virginia P. Sybert MD, editor. Genetic skin disorders. Oxford University Press: 1997. p. 352-5.
[Google Scholar]
|
| 6. |
Hinek A, Smith AC, Cutiongco EM, Callahan JW, Gripp KW, Weksberg R. Decreased elastin deposition and high proliferation of fibroblasts from Costello syndrome are related to functional deficiency in the 67-kD elastin-binding protein. Am J Med Genet 2000;66:859-72.
[Google Scholar]
|
| 7. |
Kerr B, Delrue MA, Sigaudy S, Perveen R, Marche M, Burgelin I, et al . Genotype-phenotype correlation in Costello syndrome: HRAS mutation analysis in 43 cases. J Med Genet 2006;43:401-5.
[Google Scholar]
|
| 8. |
Estep AL, Tidyman WE, Teitell MA, Cotter PD, Rauen KA. HRAS mutations in Costello syndrome: Detection of constitutional activating mutations in codon 12 and 13 and loss of wild-type allele in malignancy. Am J Med Genet A 2006;140:8-16.
[Google Scholar]
|
| 9. |
Johnson JP, Golabi M, Norton ME, Rosenblatt RM, Feldman GM, Yang SP, et al . Costello syndrome: phenotype, natural history, differential diagnosis and possible cause. J Pediatr 1998;133:441-8.
[Google Scholar]
|
| 10. |
Levaillant JM, Gιrard-Blanluet M, Holder-Espinasse M, Valat-Rigot AS, Devisme L, Cavι H, et al . Prenatal phenotypic overlap of Costello syndrome and severe Noonan syndrome by tri-dimensional ultrasonography. Prenat Diagn 2006;26:340-4.
[Google Scholar]
|
| 11. |
White SM, Graham JM Jr, Kerr B, Gripp K, Weksberg R, Cytrynbaum C, et al . The adult phenotype in Costello syndrome. Am J Med Genet A 2005;136:128-35.
[Google Scholar]
|
Fulltext Views
4,213
PDF downloads
2,899





![[Figure - 1]](#fig_ijdvl_2007_73_6_406_37059_1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2007_73_6_406_37059_2.jpg){kind=link}
![[Figure - 3]](#fig_ijdvl_2007_73_6_406_37059_3.jpg){kind=link}
![[Figure - 4]](#fig_ijdvl_2007_73_6_406_37059_4.jpg){kind=link}