Translate this page into:
Degos disease-like presentation in systemic lupus erythematosus
Correspondence Address:
Najeeba Riyaz
Department of Dermatology and Venereology, Medical College, Calicut
India
| How to cite this article: Riyaz N, Saleem R, Shafeeq R. Degos disease-like presentation in systemic lupus erythematosus. Indian J Dermatol Venereol Leprol 2011;77:219-221 |
Sir,
Degos disease (DD) or malignant atrophic papulosis or Kohlmeier-Degos-Delort-Tricort syndrome was first described in 1941 by Kohlmeier, and Robert Degos recognized it as a distinct entity in 1942. [1],[2] This rare disorder is probably an endovasculitis affecting small- and medium-sized arteries leading to tissue infarction mainly in the skin, gastrointestinal system, and central nervous system. It is characterized by porcelain white atrophic papules with infarctive lesions. Variants of DD with only cutaneous involvement and association with collagen vascular disease have been described. [1],[3] Hence, DD is considered as a distinctive reaction pattern rather than a specific disease.[4] We report a patient with the benign cutaneous form of DD whose immunological profile was suggestive of systemic lupus erythematosus (SLE).
A 32-year-old male presented with multiple erythematous papules and nodules mainly over the back of trunk and proximal extremities of 2 weeks duration. These lesions rapidly developed necrosis at the center which eventually healed leaving behind porcelain-white, atrophic scars within a week. He gave history of photosensitivity and erythema over the malar area.
Cutaneous examination revealed a faint malar rash with multiple skin color and erythematous papules and nodules of size 0.5-1 cm over the trunk. Some papules had central necrosis. Multiple round and linear atrophic porcelain white scars were seen over both arms and forearms, some having a rim of erythema [Figure - 1]. Atrophic telangiectatic lesions were seen on the tips of thumb, index, and middle fingers.
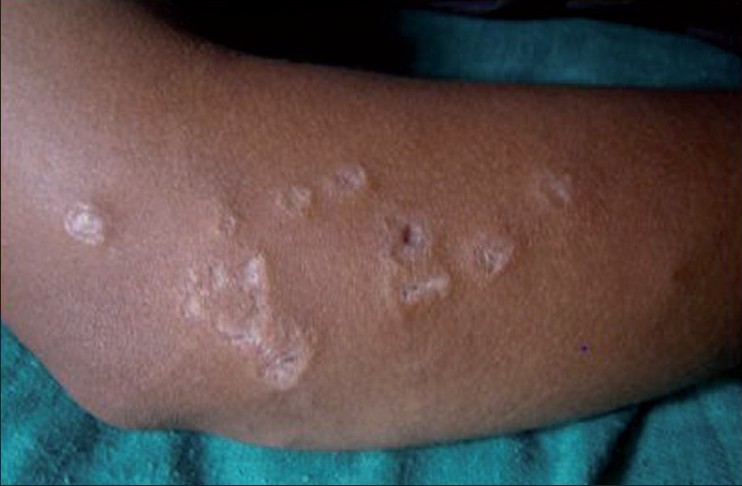 |
| Figure 1: Papule with porcelain white atrophic centre |
Histopathology from an erythematous papule with central depression revealed dermal edema, mucin deposition and lymphocytic vasculitis consistent with DD [Figure - 2]. A biopsy from a nodule showed basal cell degeneration, interface dermatitis, and periappendageal and perivascular infiltrate suggestive of SLE. The immunological profile revealed positive ANA, RA factor, anti-Sm, anti-u 1 -RNP, anti-SS-A, and antiplatelet antibodies. With these findings we came to a diagnosis of benign cutaneous form of DD with SLE. He was treated with hydroxychloroquine and low dose aspirin.
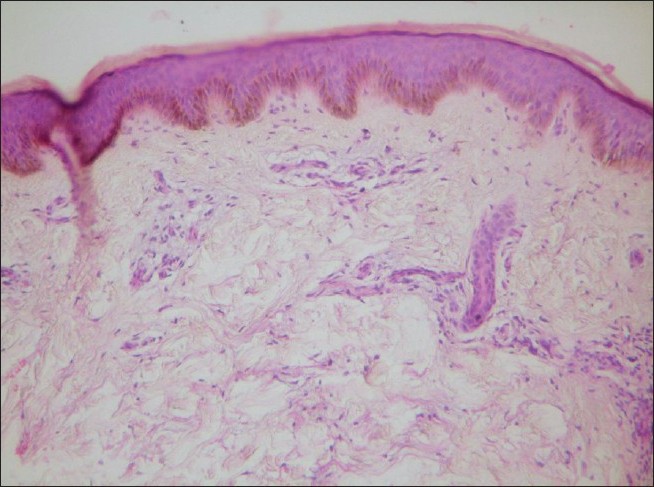 |
| Figure 2: Dermal edema and mucin deposition with lymphocytic vasculitis, consistent with Degos disease (H and E, ×40) |
DD is an uncommon condition characterized by asymptomatic papules which over a few days to weeks develop porcelain white depressed scars with rosy rim partly due to delicate telangiectases. A fatal outcome can occur within 1-2 years due to gastrointestinal and central nervous systems involvement.
Our patient had only cutaneous features suggestive of the benign cutaneous form of DD. Harvell et al have reported cases with only cutaneous involvement. About 4-15% of patients could have a benign course. Our patient also had malar rash, photosensitivity, positive ANA, and anti-Sm antibody, and past history of thrombocytopenia, suggestive of SLE. A review of the literature showed that until 2000, 16 patients with typical lesions of DD seemed to have other diseases concurrent with it, mainly collagen vascular diseases,[4] of which 11 had systemic lupus erythematosus and one each had rheumatoid arthritis, dermatomyositis, progressive systemic sclerosis, drug-induced immunosuppression, and AIDS. Dubin [5] et al described a 29-year-old man with clinical and histological features of DD and immunological features suggestive of SLE similar to our case. Black reported two females with SLE who developed atrophic papules of DD.[1] Histopathology of the early papule had shown abundant mucin which was similar to our finding. A biopsy from another lesion was suggestive of SLE. Doutre et al[3] have also reported similar findings.
According to Ball et al,[4] DD is a distinctive morphologic pattern induced by many different factors, rather than a disease with a single specific cause.
Since our patient had only cutaneous lesions and no systemic involvement, he was treated with hydroxychloroquine.
This case is reported for its exceptionally interesting and rare presentation with a varied immunological profile. Patients with skin lesions typical of DD should be assessed for other associated diseases like SLE, other collagen vascular diseases, and immunosuppressive disorders.
| 1. |
Black MM. Malignant atrophic papulosis (Degos disease). Int J Dermatol 1976;5:405-11
[Google Scholar]
|
| 2. |
Scheinfeld NS. Malignant atrophic papulosis. Clin Exp Dermatol 2007;32:483-7.
[Google Scholar]
|
| 3. |
Doutre MS, Beylot C, Bioulac P, Busquet M, Conte M. Skin lesions resembling malignant atrophic papulosis in lupus erythematosus. Dermatologica 1987;175:45-6.
[Google Scholar]
|
| 4. |
Ball E, Newburger A, Ackerman AB. Degos' disease: A distinctive pattern of disease chiefly of lupus erythematosus and not a specific disease per se. Am J Dermatopathol 2003;25:308-20.
[Google Scholar]
|
| 5. |
Dubin HV, Stawiski MA. Systemic lupus erythematosus resembling malignant atrophic papulosis. Arch Int Med 1974;134:321-3.
[Google Scholar]
|
Fulltext Views
3,965
PDF downloads
1,917





![[Figure - 1]](#fig_ijdvl_2011_77_2_219_77477_f1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2011_77_2_219_77477_f2.jpg){kind=link}