Translate this page into:
Detection of the STS gene in a family with X-linked recessive ichthyosis
2 Shandong Academy of Medical Science, Jinan, Shandong Province, China
3 Shandong Provincial Medical Center for Dermatovenereology, Jinan, Shandong Province; Shandong Provincial Hospital for Skin Diseases, Jinan, Shandong Province, China
4 Shandong Provincial Institute of Dermatology and Venereology, Shandong Academy of Medical Sciences; Shandong Provincial Key Lab for Dermatovenereology, Jinan, Shandong Province; Shandong Provincial Medical Center for Dermatovenereology; Shandong Provincial Hospital for Skin Diseases, Jinan, Shandong Province, China
Correspondence Address:
Furen Zhang
Shandong Provincial Institute of Dermatology and Venereology, Jingshi Road, Jinan, Shandong Province
China
| How to cite this article: Wang N, An K, Liu H, Fu X, Yu G, Yu Y, Tian H, Zhang F. Detection of the STS gene in a family with X-linked recessive ichthyosis. Indian J Dermatol Venereol Leprol 2013;79:268 |
Sir,
X-linked recessive ichthyosis (XLRI, OMIM: 308100) is a genetic disorder of keratinization characterized by dark, adhesive, polygonal, and regular scales of skin. Corneal opacity could also be observed in 10-15% of affected individuals. [1] XLRI usually occurred at birth or within 3 months after birth, and affects roughly 1:2000 to 1:6000 males. In 1978, the steroid sulphatase enzyme (STS) deficiency was identified to be responsible for the onset of XLRI. [2] The majority of XLRI patients have complete deletions of the STS gene. Here we performed a mutation analysis of the STS gene in a Chinese XLRI family.
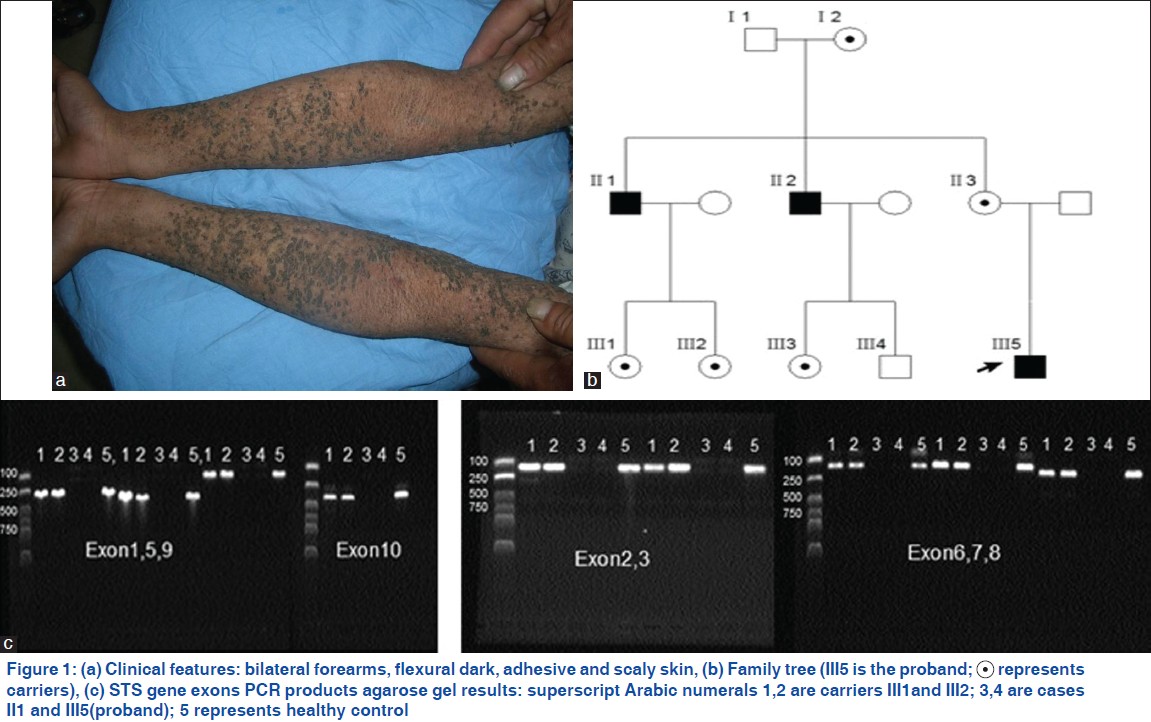
The study protocol was approved by ethical committee of Shandong provincial institute of dermatovenereology. All the family members were recruited with written informed consent. The proband (ǁǀ5) is a 9 year-old boy with generalized dry and grey brown scales, without involvement of the palms, soles and face, and no palmar hyperlinearity, accompanied by severe itch [Figure - 1]a. The symptoms occurred after 1 month of his birth and could get clinical ease during the summer. There was no history of other diseases (without corneal opacity and cryptorchidism) in his family and the same symptoms had also been observed in his two maternal uncles [Figure - 1]b. According to his clinical manifestations and family genetic model, XLRI could be diagnosed.
 |
| Figure 1: |
DNA samples were isolated from peripheral-blood leukocytes via standard protocol of QIAquick gel extraction kit (QIAGEN, Valencia, CA, USA). Polymerase chain reaction (PCR) was used in this study. STS gene was amplified by PCR with primers described by Margarita Valdes-Flores, et al.,[3] previously. All primer pairs were confirmed to be specific for the STS gene on chromosome X by database queries (using BLAST). The PCR reaction was conducted on a Veriti thermal cycler (Applied Biosystems, Foster City, CA, USA) in a total volume of 25 μL. The PCR products were tested in an agarose gel at 1% concentration. All the STS exons failed to be detected in the three XLRI patients, while clear bands of fragments in three female carriers and 100 healthy controls could be observed [[Figure - 1]c showed a part of results]. Three repeated experiments of all the exons were performed for the confirmation of results.
XLRI is an inborn error of metabolism inherited as an X-linked recessive trait, only seven female cases have been reported to date. There is no noticeable racial or geographic predilection. [1] As the causal gene of XLRI, the deficiency of STS could result in the accumulation of cholesterol sulfate in the membranes of stratum corneum cells, while cholesterol sulfate has been proven to play a role in membrane integrity and normal desquamation within the stratum corneum. [4] In female carriers of XLRI, STS activity decreased, who can have dry skin in winter without other recognizable phenotypes. And it has also been reported that a small number of cases may be the consequence of other genetic alterations of the X chromosome not directly linked to the STS gene, which showed some genetic heterogeneity. [5]
By reviewing the literatures, we found that approximately 80-90% of cases of XLRI were due to the totally absence of STS gene. Only 8 partial mutations and 14 point mutations of STS gene have been reported, most of which located at the 3′ end of the STS gene, approximately 86% (19/22). However, the clinical severity of XLRI is equal to the patients caused by the complete deletion of STS gene, which indicates the importance of the 3′ end of the STS gene. In conclusion, our XLRI family with totally absence of STS gene was investigated by direct PCR. The analysis for STS gene is still the gold standard for diagnosis of XLRI and the identification of molecular basics of the family will increase the understanding of the genetics of the STS gene and be useful for the prenatal consultation of this family.
| 1. |
Hernández-Martín A, González-Sarmiento R, De Unamuno P. X-linked ichthyosis: An update. Br J Dermatol 1999;141:617-27.
[Google Scholar]
|
| 2. |
Koppe G, Marinkovic-Ilsen A, Rijken Y, De Groot WP, Jöbsis AC. X-linked ichthyosis. A sulphatase deficiency. Arch Dis Child 1978;53:803-6.
[Google Scholar]
|
| 3. |
Valdes-Flores M, Kofman-Alfaro SH, Vaca AL, Cuevas-Covarrubias SA. Mutation report: A novel partial deletion of exons 2-10 of the STS gene in recessive X-linked ichthyosis. J Invest Dermatol 2000;114:591-3.
[Google Scholar]
|
| 4. |
Yen PH, Allen E, Marsh B, Mohandas T, Wang N, Taggart RT, et al. Cloning and expression of steroid sulfatase cDNA and the frequent occurrence of deletions in STS deficiency: Implications for X-Y exchange. Cell 1987;49:443-54.
[Google Scholar]
|
| 5. |
Robledo R, Melis P, Schillinger E, Casciano I, Balazs I, Rinaldi A, et al. X-linked ichthyosis without STS deficiency: Clinical, genetical, and molecular studies. Am J Med Genet 1995;59:143-8.
[Google Scholar]
|
Fulltext Views
2,857
PDF downloads
1,827





![[Figure - 1]](#fig_ijdvl_2013_79_2_268_107669_f1.jpg){kind=link}