Translate this page into:
Easy bruising due to giant platelet (possibly MYH9-related/Sebastian) syndrome
2 Department Of Pathology, Medical College and Hospital, Kolkata, India
Correspondence Address:
Indrashis Podder
M.D. (Dermatology), Medical College and Hospital, Kolkata Block AD-71, Salt Lake City, Kolkata - 700 064, West Bengal
India
| How to cite this article: Podder I, Saha M, Das A, Gharami RC. Easy bruising due to giant platelet (possibly MYH9-related/Sebastian) syndrome. Indian J Dermatol Venereol Leprol 2015;81:633-635 |
Sir,
The easy bruising syndrome is a condition characterized by recurrent bruising on trivial trauma despite a normal coagulation profile.[1] Some of these cases, collectively called inherited giant platelet disorders, may show large platelets, thrombocytopenia and neutrophilic inclusions on peripheral blood smear examination. Sebastian syndrome is one of the rarer inherited giant platelet disorders. We were unable to find any previous reports from India.
A 5-year-old girl was brought to our dermatology out-patient department at the Medical College and Hospital, Kolkata with a complaint of recurrent bruising with trivial trauma for 7 months. These bruises occurred mostly on her lower limbs and were transient. There was no history of epistaxis or gingival bleeding. No history of physical abuse could be elicited. Her father and sister also had similar complaints. She was born to non-consanguineous parents and her development had been normal. There was no history suggestive of hearing loss, visual impairment (cataract) or nephritis in the patient or in her family. She had never received platelet or red blood cell transfusions.
Cutaneous examination revealed well-defined, ecchymotic patches on the lateral [Figure - 1] and anterior aspects of the mid-thigh, around 4 cm × 3 cm in size. Hair, nails and mucosa were normal. Systemic examination including audiological and ophthalmological evaluation were non-contributory; there was no lymphadenopathy or organomegaly. Hematological investigations were normal except for mild thrombocytopenia (platelet count 90 × 103/cu.mm). The coagulation profile (bleeding time, partial thromboplastin time, prothrombin time) and platelet aggregation studies (turbidometric method) using adrenaline, ristocetin sulphate, collagen, arachidonic acid and adenosine as agonist or aggregating agents were also within normal limits thus ruling out the Von-Willebrand subtypes of diseases. Peripheral blood smear examination showed large platelets (macrothrombocytes) [Figure - 2] and non-specific cytoplasmic inclusions within neutrophils [Figure - 3]. It was evident that these inclusions were not staining artifacts as they were spread unevenly in most of the neutrophils, (and not uniformly as expected with artifacts). They also appeared different from the blue-black toxic granules seen in infection and were not associated with cytoplasmic vacuoles as in the latter. The patient's father and sister also had mild thrombocytopenia with normal coagulation studies and peripheral blood smears. A diagnosis of Sebastian syndrome was considered and the patient advised regular follow-up.
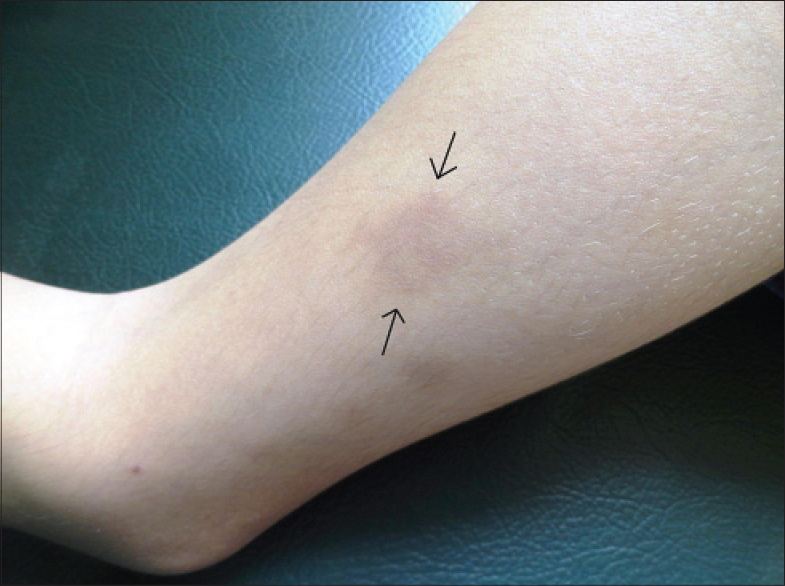 |
| Figure 1: Well-defined ecchymotic patch on the lower leg |
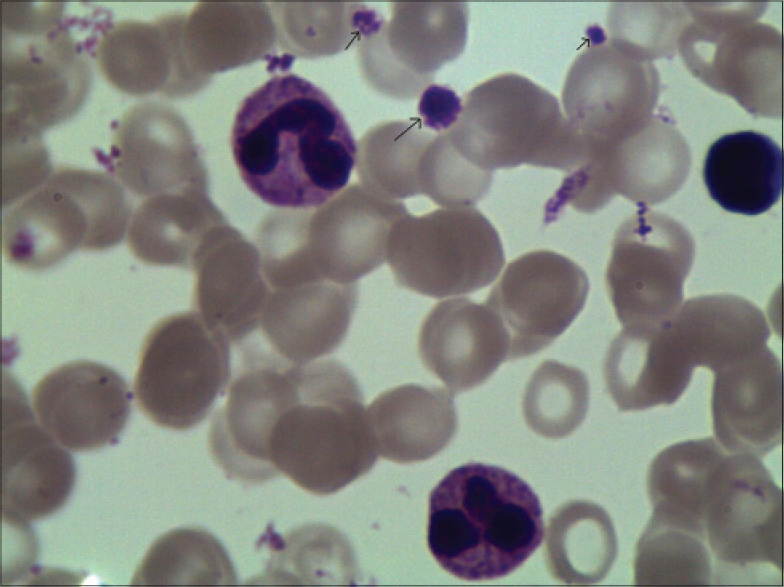 |
| Figure 2: Peripheral blood smear showing platelets (arrows), a giant platelet can be seen in the centre (arrow) |
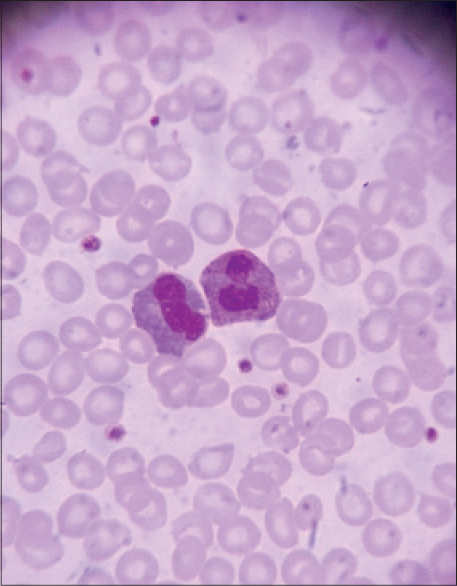 |
| Figure 3: Peripheral blood smear showing neutrophilic inclusions |
The easy bruising syndrome can be broadly classified into Type I with normal platelets and Type II with platelet abnormalities. There may be anti-platelet antibodies in both, hinting at an autoimmune etiology.[2]
Physical trauma appears to be the most important cause for Type I easy bruising, which was ruled out in our case due to an absence of a history of significant trauma, the presence of petechiae and a positive family history.
Type II easy bruising syndrome comprises inherited disorders associated with fewer and larger platelets (macrothrombocytopenia); these disorders are clubbed together as inherited giant platelet disorders [Table - 1]. Two of these (May–Hegglin anomaly and Sebastian syndrome), associated with abnormal neutrophilic inclusions occur due to variable expression of the MYH-9 gene.[3] May–Hegglin anomaly consists of Dohle-like neutrophilic inclusions.[1] Dohle bodies are small, round to oval, pale-blue to grey structures usually found at the periphery of neutrophils, ultrastructurally consisting of ribosomes and endoplasmic reticulum.[4] In Sebastian syndrome on the other hand, as in our case, non-specific inclusions are found in the cytoplasm of neutrophils. Other MYH9-related disorders are Fechtner syndrome (deafness, cataract, nephritis) and Epstein syndrome (nephritis, sensorineural hearing loss).[5] The MYH9-related disorders are currently believed not to represent distinct entities but rather varied expressions of a single illness. They may be asymptomatic or present with easy bruising as in our case. Rarely, bleeding from other sites (nose, gums, vagina) may be present.[6]
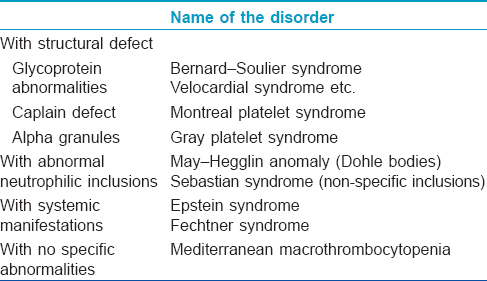
The main differential diagnoses ruled out in our case were acute idiopathic thrombocytopenia (acute onset, usually with bleeding from other sites and a prolonged bleeding time), various types of coagulation disorders (altered coagulation profile), child abuse (ruled out by a careful history and the absence of suggestive bruises elsewhere) and other inherited giant platelet disorders [Table - 1]. No systemic involvement was found in our case ruling out Fechtner and Epstein syndromes. The non-specific neutrophilic inclusions in our case pointed towards Sebastian syndrome.
Sebastian syndrome is an extremely rare autosomal dominant disorder, first described by Greinacher et al. in 1990.[7] Since then, only a handful of cases have been reported, including one South American [8] and six Japanese families.[9] It is characterized by the presence of thrombocytopenia with giant platelets and non-specific neutrophilic inclusions. Apart from easy bruising, there is usually no symptom. The pathogenesis of this condition is unclear though unusual expression of glycophorin A on the surface of platelets has been proposed.[10] There is no known specific treatment for Sebastian syndrome. Platelet transfusion is the treatment of choice in the rare situation of excessive bleeding. The role of 1-deamino-8-arginine vasopressin and spenectomy is controversial.[5]
Considering the history, clinical features and hematological picture in this case, we arrived at the diagnosis of an MYH9-related syndrome, most likely Sebastian syndrome.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
| 1. |
James WD, Elston DM, Berger TG. Andrews' Diseases of the skin: Clinical Dermatology. 11th ed. United Kingdom: Saunders Elsevier; 2006p. 820-1.
[Google Scholar]
|
| 2. |
Lackner H, Karpatkin S. On the “easy bruising” syndrome with normal platelet count. A study of 75 patients. Ann Intern Med 1975;83:190-6.
[Google Scholar]
|
| 3. |
Seri M, Pecci A, Di Bari F, Cusano R, Savino M, Panza E, et al. MYH9-related disease: May-Hegglin anomaly, Sebastian syndrome, Fechtner syndrome, and Epstein syndrome are not distinct entities but represent a variable expression of a single illness. Medicine (Baltimore). 2003;82:203-15.
[Google Scholar]
|
| 4. |
Bain BJ. Blood cell morphology in health and disease. In Practical Haematology by Dacie and Lewis. In: Bain BJ, Bates I, Laffan MA, Lewis SM, editors. United Kingdom: Elsevier Saunders. 2012. p. 69-101.
[Google Scholar]
|
| 5. |
Mhawech P, Saleem A. Inherited Giant Platelet Disorders: Classification and Literature review. Am J Clin Pathol 2000;113:176-90.
[Google Scholar]
|
| 6. |
Wimalachandra MC, Prabashika MG, Gooneratne LV. Thrombocytopenia, giant platelets and Leukocyte inclusion bodies in a patient presenting with vaginal bleeding. Indian J Pathol Microbiol 2012;55:423-4.
[Google Scholar]
|
| 7. |
Greinacher A, Nieuwenhuis HK, White JG. Sebastian platelet syndrome: A new variant of hereditary macrothrombocytopenia with leucocyte inclusions. Blut 1990;61:282-8.
[Google Scholar]
|
| 8. |
Balderramo DC, Ricchi BN, Marun SG, Scaliter G, Alonso M. Sebastian: Report of the first case in a South American family. Haematologica 2007;88:e74-e7.
[Google Scholar]
|
| 9. |
Kunishima S, Kojima T, Matsushita T, Tanaka T, Tsurusawa M, Furukawa Y, et al. Mutations in the NMMHC-A gene cause autosomal dominant macrothrombocytopenia with leukocyte inclusions (May-Hegglin anomaly/Sebastian syndrome). Blood 2001;97:1147-9.
[Google Scholar]
|
| 10. |
Gilman AL, Sloand E, White JG, Sacher R. A novel hereditary thrombocytopenia. J Pediatr Hematol Oncol 1995;17:296-305.
[Google Scholar]
|
Fulltext Views
4,141
PDF downloads
367





![[Figure - 1]](#fig_ijdvl_2015_81_6_633_168354_f1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2015_81_6_633_168354_f2.jpg){kind=link}
![[Figure - 3]](#fig_ijdvl_2015_81_6_633_168354_f3.jpg){kind=link}
![[Table - 1]](#tbl_ijdvl_2015_81_6_633_168354_t4.jpg){kind=link}