Translate this page into:
Ectodermal dysplasia-skin fragility syndrome
Correspondence Address:
Vijay S Adhe
Department of Dermatology, OPD-117, 1st floor, Old OPD Building, Seth GS Medical College and King Edward Memorial Hospital, Parel, Mumbai - 400 012
India
| How to cite this article: Adhe VS, Dongre AM, Khopkar US. Ectodermal dysplasia-skin fragility syndrome. Indian J Dermatol Venereol Leprol 2011;77:503-506 |
Abstract
Ectodermal dysplasia-skin fragility (EDSF) syndrome is a rare and first described inherited disorder of desmosomes. It occurs due to loss-of-function mutations in PKP1 gene resulting in poorly formed desmosomes and loss of desmosomal and epidermal integrity. We report a case of a 2-year-old Indian male child who presented with palmoplantar hyperkeratosis with fissuring, short, sparse, and easily pluckable scalp hair, nail dystrophy, and multiple erosions over the skin. Skin biopsy showed epidermal hyperplasia with widening of intercellular spaces. His developmental milestones were delayed but intelligence was normal. Echocardiography, X-ray chest, and electrocardiogram were normal. Very few cases of this syndrome have been reported in the literature. We consider this as the first case report from India.Introduction
Ectodermal dysplasia-skin fragility (EDSF) syndrome also known as McGrath syndrome is a recently described, rare autosomal recessive genodermatosis, which results from loss-of-function mutations in plakophilin 1 (PKP1), a component of desmosome cell-cell junctions. McGrath et al. [1] described the first case and coined the term EDSF syndrome in 1997. Since then 10 cases of this syndrome have been reported from different parts of the world. [1] Clinically, loss of plakophilin 1 leads to skin fragility, blisters and erosions following minor trauma, palmoplantar keratoderma with painful cracking, hair abnormality, and nail dystrophy. It is now considered as a specific suprabasal form of epidermolysis bullosa simplex.
Case Report
A 2-year-old male child presented to our outpatient department of dermatology with hard and fissured skin of palms and soles, lusterless, sparse and easily pluckable scalp hair, deformed nails since birth and peeling of skin on minor trauma since 3 months of age. The child was born of second degree consanguineous marriage as full-term normal delivery and there was no history of other family members affected with similar skin disease. At birth there were no blisters or erosions on the skin. Erosions and peeling of the skin were first noted on the extremities at the age of 3 months. On further probing there was a history of delayed developmental milestones. There was no history of decreased sweating.
Cutaneous examination showed diffuse hyperkeratosis with fissuring on both the soles with relative sparing of the insteps [Figure - 1]. Erythema, hyperlinearity, and mild scaling were noted on both the palms. Nails showed leukonychia, horizontal ridging, subungual hyperkeratosis, and distal dystrophy [Figure - 2] and [Figure - 3]. Scalp hair was thin, lusterless, short, curly, and easily pluckable while eyelashes were long, dark, and curly [Figure - 4]. Multiple small erosions were present on face, including perioral area, dorsa of fingers, toes, and extremities [Figure - 2], [Figure - 3] and [Figure - 4]. Nikolsky′s sign was negative. Oral mucosa was normal and there were no dental anomalies.
 |
| Figure 1: Plantar hyperkeratosis with fissuring |
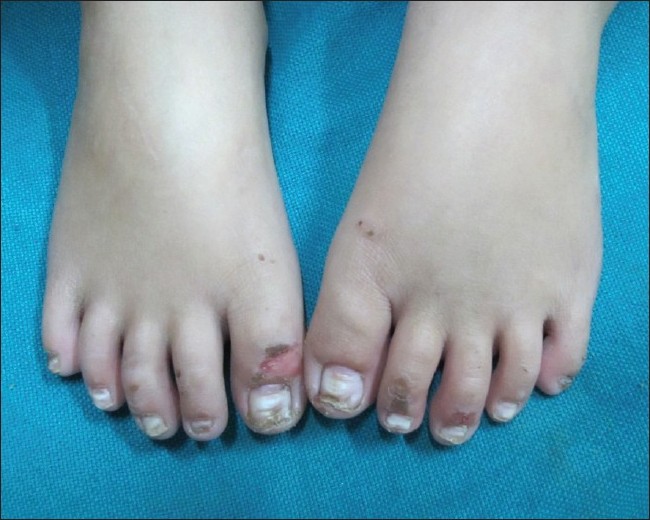 |
| Figure 2: Toe nails showing horizontal ridging, leukonychia, distal dystrophy with erosions on dorsa of toes |
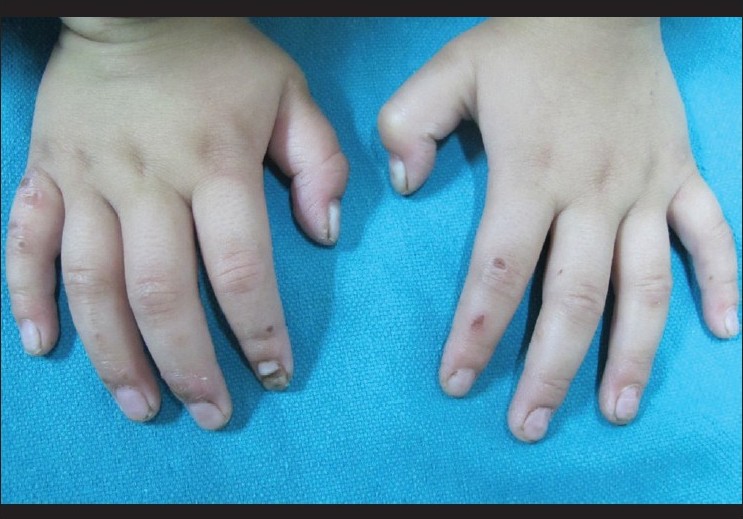 |
| Figure 3: Multiple small erosions on dorsa of hands with nail dystrophy |
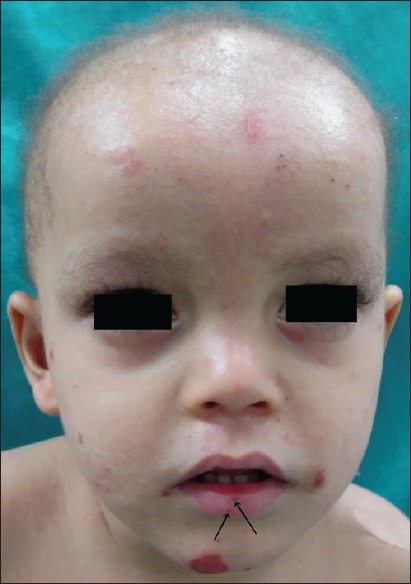 |
| Figure 4: Thin, lusterless, short, sparse scalp hair with erosion on forehead, lower lip (arrow), and perioral area |
His routine blood investigations, ECG, Roentgenogram of chest and 2-D Echo were normal. Skin biopsy from erosion showed acanthosis with parakeratosis. Widening of keratinocyte intercellular spaces and a few acantholytic cells were noted in the spinous layer with intact basal layer [Figure - 5] and [Figure - 6]. Hair analysis on light microscopy and under polarization didn′t reveal any abnormality. Trichogram showed normal anagen to telogen ratio.
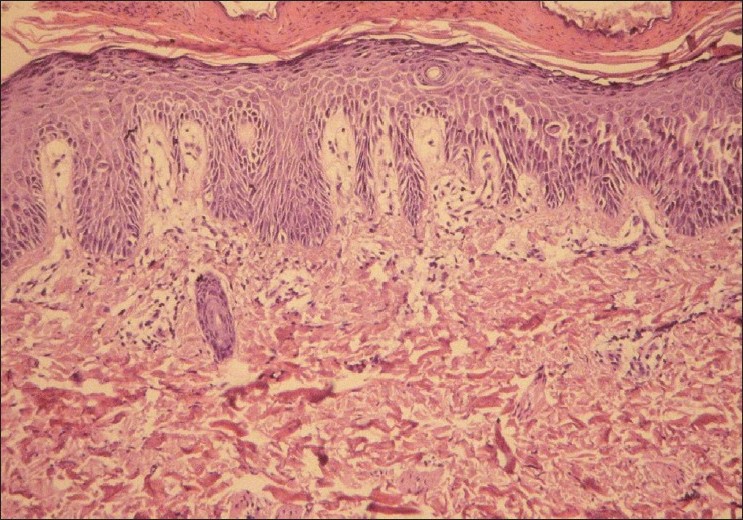 |
| Figure 5: Hyperkeratosis and acanthosis with widening of intercellular spaces of spinous layer (H and E, ×100) |
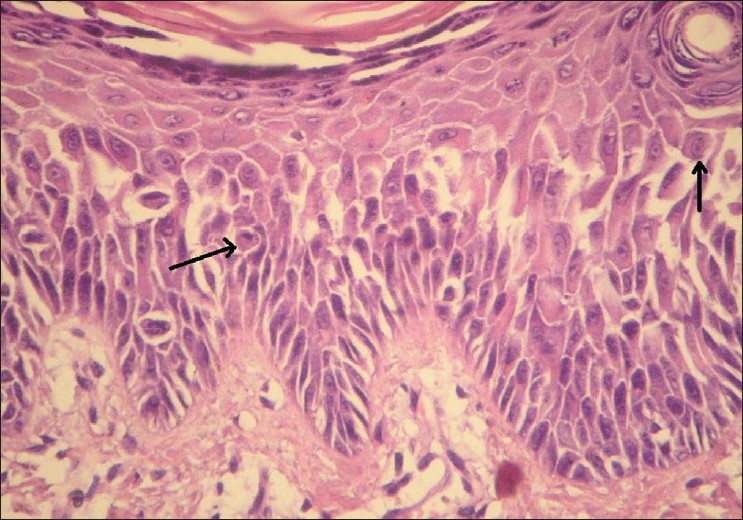 |
| Figure 6: Higher magnification showing spinous cell acantholysis (arrow) with intact basal layer (H and E, ×400) |
Discussion
EDSF syndrome is a recently described rare autosomal recessive genodermatosis affecting skin, nails, and hair (MIM 604536). It is the first inherited disorder of desmosomes.
EDSF syndrome was first described by McGrath and colleagues in 1997, in a 6-year-old boy who presented with blistering on soles at birth with short, sparse hair, and dystrophic nails along with skin fragility, trauma-induced erosions, and blisters. [2] Since then 10 cases of this rare syndrome have been reported from various parts of the world showing similar clinical abnormalities.
EDSF results from loss of function mutations in the PKP1 gene that encodes for a protein, plakophilin 1 (PKP1), which is a member of the armadillo family of structural and signaling proteins. It is the component of desmosomal plaque, which links desmosomal cadherins (desmogleins and desmocollins) to the keratins that make up the intermediate filament. Thus, it contributes to the mechanical integrity and calcium stability of desmosomes. [3],[4]
PKP1 is expressed throughout the epidermis, particularly in suprabasal cells and also in the outer root sheath of hair follicles. [5] Two isoforms of PKP1, 1a and 1b have been described. [6]
PKP1a is expressed in both desmosomes and nuclei, whereas PKP1b is expressed only in nuclei. Currently, the specific biologic significance of the two isoforms is unknown. [7]
Various features, such as skin fragility, alopecia, palmoplantar keratoderma, hypohidrosis, nail dystrophy, and cheilitis, have been described in patients with EDSF syndrome. Of these, skin fragility, alopecia, and palmoplantar keratoderma are the most consistent features. Uncommon features such as pruritus, growth retardation, and lens abnormalities have also been reported. Hypohidrosis, cheilitis, pruritus, lens abnormalities, and dental caries reported in some cases of EDSF syndrome were absent in our case. Classical features like perioral fissuring and cheilitis are not reported in many cases. Also a case of EDSF syndrome without palmoplantar keratoderma has been described in literature. [1]
Skin fragility and blistering following trauma in children also occurs in epidermolysis bullosa. The third International Consensus Meeting on the Diagnosis and Classification of EB has classified EDSF syndrome as a specific suprabasal form of epidermolysis bullosa simplex. [8] Cardiomyopathy seen in other inherited desmosomal disorders, such as Naxos or Carvajal syndrome, is not seen in patients with EDSF syndrome as PKP1 is not expressed in the heart. [6]
Cases with EDSF syndrome like clinical presentation, but with mutations in genes other than PKP1, that is, desmoplakin and plakoglobin, have been reported in the literature. [9],[10] Although diseases caused by mutations in desmosomal proteins share various clinical features, such as keratoderma and alopecia, EDSF syndrome is the only condition which shows skin fragility as a prominent feature. [11]
Other cutaneous disorders with skin fragility or blistering presenting in the early life include porphyria, Kindler syndrome but are associated with photosensitivity, scarring, atrophy, and sclerodermoid changes. Epidermolysis bullosa aquisita presents as bullae/erosions on trauma prone areas, mucosae, and heals with scarring and milia formation. Now EDSF syndrome is considered as a specific suprabasal form of epidermolysis bullosa simplex.
Histopathological examination of skin biopsy shows hyperkeratosis, acanthosis with widened intercellular spaces, and acantholytic keratinocytes. Along with these features dyskeratosis may be seen if plantar skin is biopsied. Although the keratinocytes have the appearance of acantholytic cells, the actual plane of cleavage or weakness lies within the desmosomal plaque (ie, inside the keratinocyte). [12]
Immunohistochemical and electron microscopic studies have demonstrated poorly developed, small desmosomes along with a reduction in the number of desmosomes in the epidermis, particularly involving the lower suprabasal layer. These findings were also noted in unaffected carriers. [13]
There is no specific treatment. Management is similar to that of epidermolysis bullosa. A prenatal diagnosis can be established by preimplantation genetic diagnosis. [14]
This is probably the first case report of EDSF syndrome from India.
| 1. |
Ersoy-Evans S, Erkin G, Fassihi H, Chan I, Paller AS, McGrath JA, et al. Ectodermal dysplasia-skin fragility syndrome resulting from a new homozygous mutation, 888delC, in the desmosomal protein plakophilin1. J Am Acad Dermatol 2006;55:157-61.
[Google Scholar]
|
| 2. |
McGrath JA, McMillan JR, Shemanko CS, Runswick SK, Leigh IM, Lane EB, et al. Mutations in the plakophilin 1 gene result in ectodermal dysplasia/skin fragility syndrome. Nat Genet 1997;17:240-4.
[Google Scholar]
|
| 3. |
South AP, Wan H, Stone MG, Dopping-Hepenstal PJ, Purkis PE, Marshall JF, et al. Lack of plakophilin 1 increases keratinocyte migration and reduces desmosome stability. J Cell Sci 2003;116:3303-14.
[Google Scholar]
|
| 4. |
South AP. Plakophilin 1: An important stabilizer of desmosomes. Clin Exp Dermatol 2004;29:161-7.
[Google Scholar]
|
| 5. |
Moll I, Kurzen H, Langbein L, Franke WW. The distribution of the desmosomal protein, plakophilin 1, in human skin and skin tumors. J Invest Dermatol 1997;108:139-46.
[Google Scholar]
|
| 6. |
McGrath JA, Mellerio JE. Ectodermal dysplasia-skin fragility syndrome. Dermatol Clin 2010;28:125-9.
[Google Scholar]
|
| 7. |
Sobolik-Delmaire T, Katafiasz D, Wahl JK 3 rd . Carboxyl terminus of plakophilin 1 recruits to its plasma membrane, whereas amino terminus recruits desmoplakin and promotes desmosome assembly. J Biol Chem 2006;281:16962-70.
[Google Scholar]
|
| 8. |
Fine JD, Eady RA, Bauer EA, Bauer JW, Bruckner-Tuderman L, Heagerty A, et al. The classification of inherited epidermolysis bullosa (EB): Report of the third International Consensus Meeting on diagnosis and classification of EB. J Am Acad Dermatol 2008;58:931-50.
[Google Scholar]
|
| 9. |
Winik BC, Asial RA, McGrath JA, South AP, Boente MC. Acantholytic ectodermal dysplasia: Clinicopathological study of a new desmosomal disorder. Br J Dermatol 2009;160:868-74.
[Google Scholar]
|
| 10. |
Cabral RM, Liu L, Hogan C, Dopping-Hepenstal PJ, Winik BC, Asial RA, et al. Homozygous mutations in the 5¢ region of the JUP gene result in cutaneous disease but normal heart development in children. J Invest Dermatol 2010;130:1543-50.
[Google Scholar]
|
| 11. |
Sprecher E, Molho-Pessach V, Ingber A, Sagi E, Indelman M, Bergman R. Homozygous splice site mutations in PKP1 result in loss of epidermal plakophilin 1 expression and underlie ectodermal dysplasia/skin fragility syndrome in two consanguineous families. J Invest Dermatol 2004;122:647-51.
[Google Scholar]
|
| 12. |
Bergman R, Sprecher E. Histopathological and ultrastructural study of ectodermal dysplasia/skin fragility syndrome. Am J Dermatopathol 2005;27:333-8.
[Google Scholar]
|
| 13. |
McGrath JA, Mellerio JE. Ectodermal dysplasia-skin fragility syndrome. Dermatol Clin 2010;28:125-9.
[Google Scholar]
|
| 14. |
Fassihi H, Grace J, Lashwood A, Whittock NV, Braude PR, Pickering SJ, et al. Preimplantation genetic diagnosis of skin fragility-ectodermal dysplasia syndrome. Br J Dermatol 2006;154:546-50.
[Google Scholar]
|
Fulltext Views
5,709
PDF downloads
3,288





![[Figure - 1]](#fig_ijdvl_2011_77_4_503_82415_f1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2011_77_4_503_82415_f2.jpg){kind=link}
![[Figure - 3]](#fig_ijdvl_2011_77_4_503_82415_f3.jpg){kind=link}
![[Figure - 4]](#fig_ijdvl_2011_77_4_503_82415_f4.jpg){kind=link}
![[Figure - 5]](#fig_ijdvl_2011_77_4_503_82415_f5.jpg){kind=link}
![[Figure - 6]](#fig_ijdvl_2011_77_4_503_82415_f6.jpg){kind=link}