Translate this page into:
Familial gigantic melanocytosis
2 Department of Dermatology, NRI Medical College and General Hospital, Chinakakani, Guntur, Andhra Pradesh, India
Correspondence Address:
Kinjal D Rambhia
C/O Dr Amit Gulati, H. No. 574-Gulati Bhavan, Mukundraj Lane, Mahal, Nagpur, Maharashtra
India
| How to cite this article: Rambhia KD, Chowdhary K S, Rao G V, Khopkar US. Familial gigantic melanocytosis. Indian J Dermatol Venereol Leprol 2018;84:192-194 |
Sir,
Familial gigantic melanocytosis is a rare and peculiar familial disorder of pigmentation. It presents as hyperpigmented and hypopigmented macules occurring predominantly in sun-exposed areas and later progressing to involve the photoprotected areas. The pigmentation may simulate the pattern of arsenic poisoning, where hyperpigmentation is interspersed with rain-drop hypopigmentation. It may also mimic other pigmentary disorders such as dyschromatosis universalis hereditaria, dyschromatosis symmetrica hereditaria, Dowling–Degos disease, reticulate acropigmentation of Kitamura and Fanconi's anemia.
A 25-year-old male presented with asymptomatic pigmentation of the face, upper extremities and the “V” area of the neck since 4 years. There was no history of itching, redness or photosensitivity. He denied using any cosmetics or hair dye. It was revealed that his paternal aunt suffered from a similar pattern of pigmentation. Cutaneous examination revealed diffuse reticulate hyperpigmentation over the face, extensor aspect of upper extremities, the “V” area of neck and photo-exposed parts of the chest [Figure - 1]a. In addition, there were multiple discrete, tiny, hypopigmented macules interspersed on the pigmented areas over the upper extremities and trunk [Figure - 1]b and [Figure - 1]c. A lightening in colour of the eyelashes, eyebrows, beard and scalp hair was noted. The oral mucosa, palms, soles and nails were normal. General and systemic examination of the patient revealed no other abnormalities. Therefore, the differential diagnoses of lichen planus pigmentosus, erythema dyschromicum perstans and dyschromatosis universalis hereditaria were considered.
 |
| Figure 1a: Reticulate hyperpigmentation over the face |
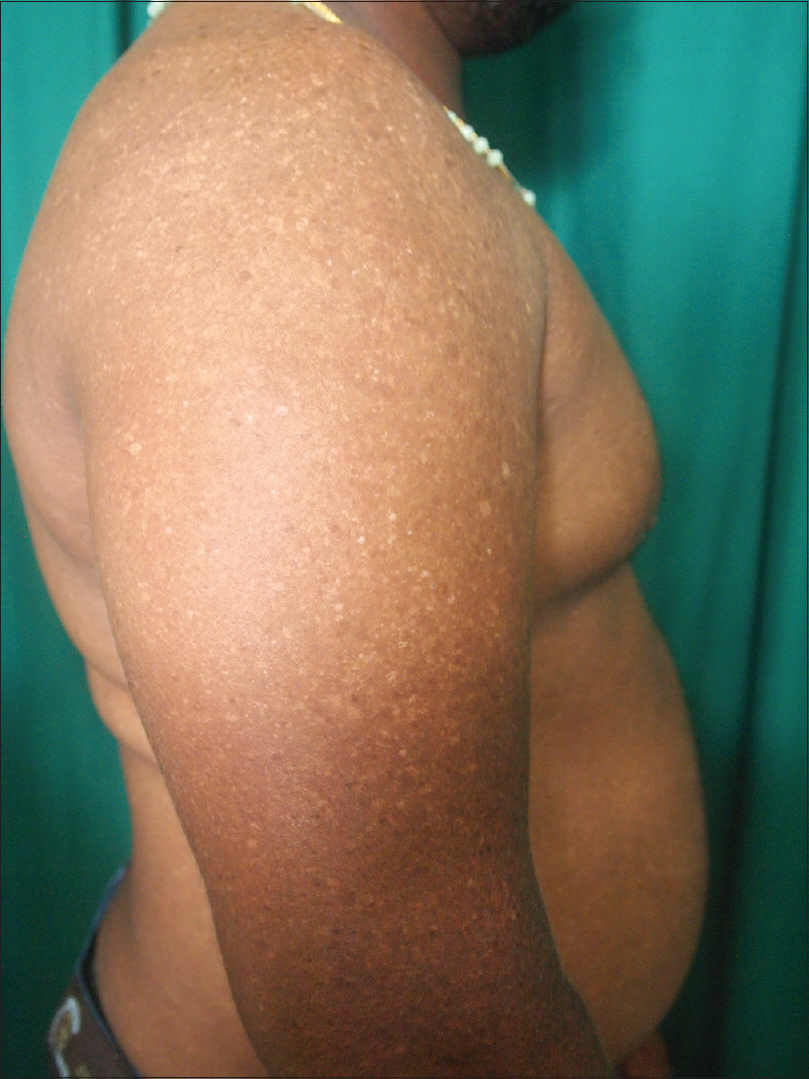 |
| Figure 1b: Hypopigmented macules over the upper extremity |
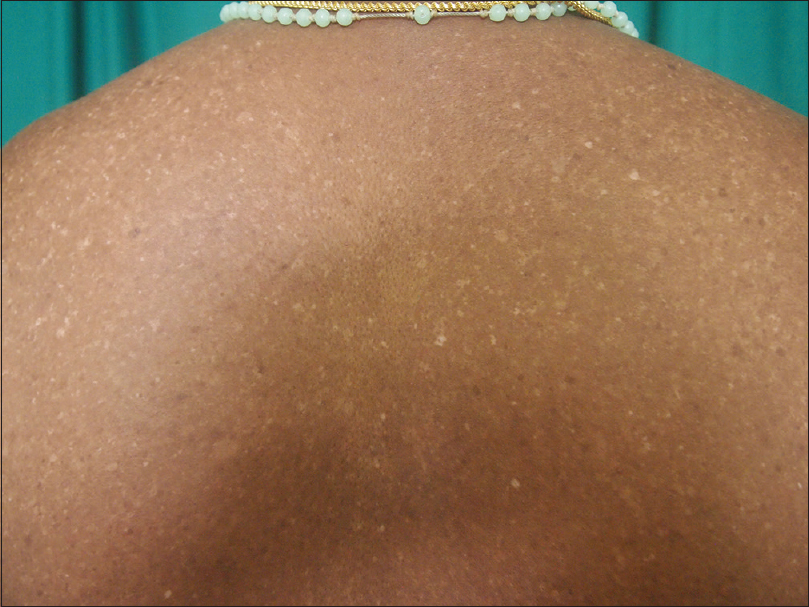 |
| Figure 1c: Hypopigmented macules on the trunk |
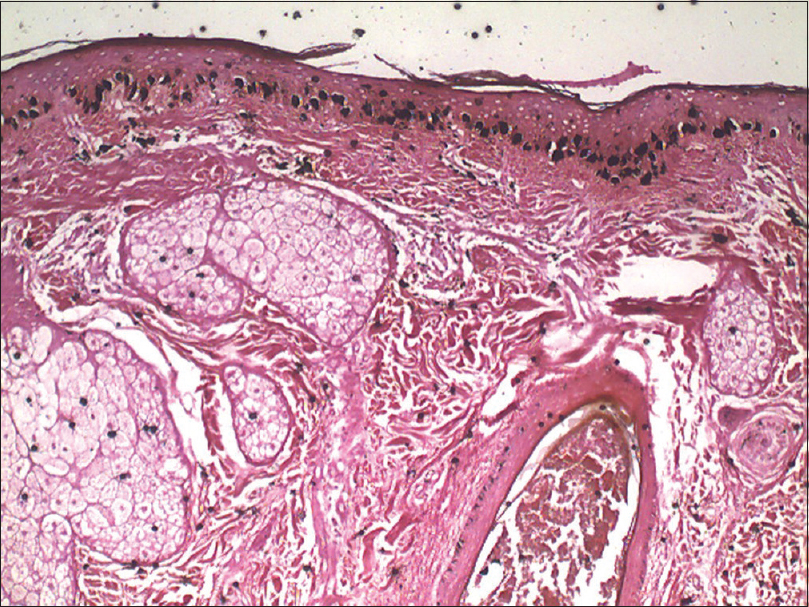
Histopathological examination of a hyperpigmented macule revealed foci of richly pigmented basal and suprabasal keratinocytes [Figure - 2]a. There were numerous huge melanocytes located in the basal layer. They were longer and larger than the normal melanocytes [Figure - 2]b. Fontana-Masson stain revealed prominent huge melanocytes that were heavily laden with melanin [Figure - 3]a and [Figure - 3]b. Thus, a diagnosis of gigantic melanocytosis was made.
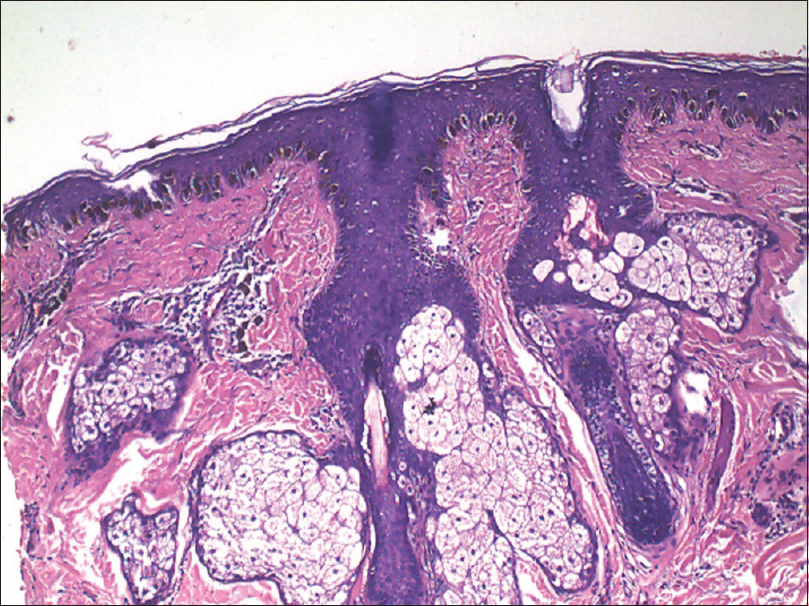 |
| Figure 2a: Increased melanocytes and pigmentation in the basal and the suprabasal layer of the epidermis (H and E, ×100) |
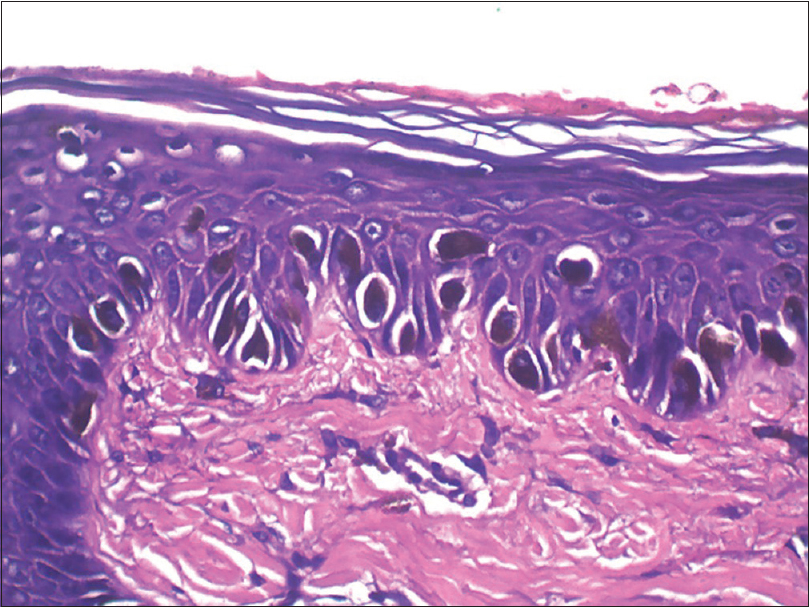 |
| Figure 2b: Numerous larger and longer melanocytes in the epidermis (H and E, ×400) |
 |
| Figure 3a: Numerous melanocytes with increased melanin (Fontana Masson, ×100) |
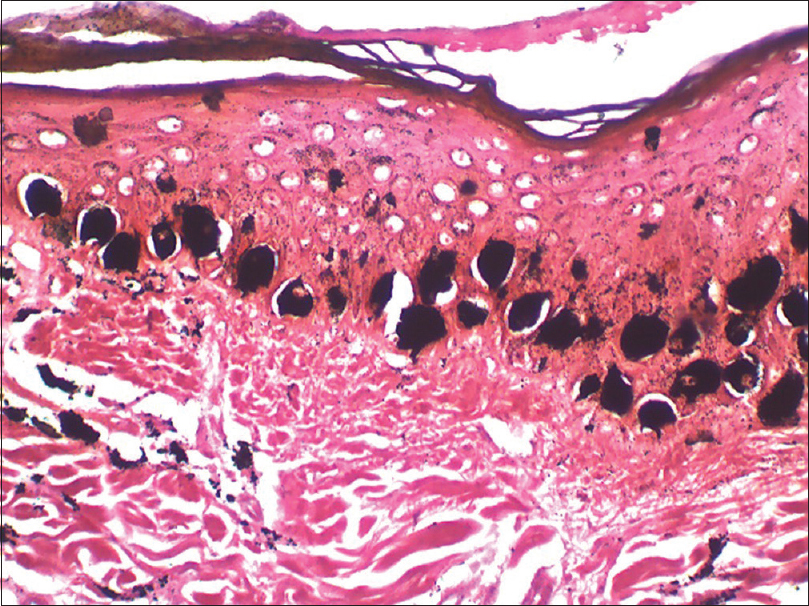 |
| Figure 3b: Larger, longer, and heavily pigmented melanocytes in the basal layer (Fontana Masson, ×400) |
Familial gigantic melanocytosis or familial melanopathy with gigantic melanocytes is a rare and peculiar familial disorder of pigmentation, first described in 1984.[1] In the cases described in literature, the onset of pigmentation was in the first year of life and the condition was more commonly seen in males. Familial occurrence was observed in majority of the described cases, which suggested it to be a hereditary disorder.
These patients clinically present with diffuse hyperpigmentation, predominantly over the photo-exposed areas; face, neck, chest, upper extremities and occasionally the trunk. There may be some hypopigmented macules interspersed within the areas of diffuse hyperpigmentation. In addition, these patients frequently have light coloured hair, eyebrows and eyelashes. There is no associated photosensitivity, itching or redness prior to the appearance of the lesions. Other sites such as palms, soles, mucosae and nails are usually spared. These patients usually do not have any systemic findings or underlying nutritional, metabolic or endocrine abnormalities.
The histopathology of familial gigantic melanocytosis is characteristic, pathognomonic and helps in clinching the diagnosis. Hematoxylin and eosin stained sections show presence of melanocytes that are larger and longer than the usual melanocytes. These gigantic melanocytes are heavily pigmented and crowded at places, whereas in other areas they appear to be sparse. Fontana-Masson silver stain highlights the giant, heavily pigmented melanocytes, crowding the basal layer of the epidermis. Electron microscopy of the hyperpigmented area demonstrates large melanosomes with large irregular nuclei having fragmented chromatin substance with an unusually wide nuclear envelope.[2]
Familial gigantic melanocytosis may clinically mimic other disorders such as lichen planus pigmentosus and disorders with reticulate pigmentation such as reticulate acropigmentation of Kitamura. However, histopathology may be helpful in arriving at the diagnosis. Lichen planus pigmentosus shows the characteristic findings of interface dermatitis with colloid bodies and melanophages in the papillary dermis. Reticulate acropigmentation of Kitamura shows increased pigmentation of the epidermis without the characteristic giant melanocytes seen in familial gigantic melanocytosis. Dowling–Degos disease shows downward proliferation of the “antler- like” rete ridges with increased pigmentation of the epidermis.
The cause of this disorder is unknown; though it is hypothesized that in genetically predisposed individuals, there is a defect in the transfer of melanosomes from the melanocytes to the surrounding keratinocytes. This results in the formation of giant, highly melanised melanosomes and paucity of melanin in the surrounding areas, causing hypopigmented macules to be interspersed within the hyperpigmented areas.
Our case was unusual due to the delayed age at presentation. In addition, there was reticulate pigmentation over the sun-exposed areas, in contrast to the diffuse hyperpigmentation in the cases originally described in literature. The reticulate hyperpigmented patches in this patient clinically simulated lichen planus pigmentosus or ashy dermatosis. However, histopathology showing gigantic melanocytes without any melanophages in the dermis was characteristic of familial melanopathy with gigantic melanocytosis.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form, the patient has given his consent for his images and other clinical information to be reported in the journal. The patient understands that the name and initials will not be published and due efforts will be made to conceal identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
| 1. |
El Darouti MA. Familial melanopathy with gigantic melanocytes. Am J Dermatopathol 1984;6 Suppl: 31-4.
[Google Scholar]
|
| 2. |
El-Darouti MA, Fawzi SA, Marzook SA, El-Eishi NH, Abdel-Halim MR, Soliman SA, et al. Familial gigantic melanocytosis. Int J Dermatol 2005;44:1010-5.
[Google Scholar]
|
Fulltext Views
2,440
PDF downloads
790





![[Figure - 1]](#fig_ijdvl_2018_84_2_192_224595_f1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2018_84_2_192_224595_f2.jpg){kind=link}
![[Figure - 3]](#fig_ijdvl_2018_84_2_192_224595_f3.jpg){kind=link}