Translate this page into:
Familial pityriasis rubra pilaris in a Chinese family caused by a novel mutation in CARD14 gene
2 BGI-Shenzhen, Shenzhen 518083, China
3 BGI-Shenzhen, Shenzhen 518083; School of Future Technology, University of Chinese Academy of Sciences, Beijing, China
4 BGI-Shenzhen, Shenzhen 518083; Xinhua College, Sun Yat-sen University, Guangdong, China
5 Xinhua College, Sun Yat-sen University; Guangzhou Institute of Obstetrics and Gynecology, Key Laboratory for Major Obstetric Diseases of Guangdong Province, Key Laboratory of Reproduction and Genetics of Guangdong Higher Education Institutes, The Third Affiliated Hospital of Guangzhou Medical University, Guangzhou 510150, Guangdong, China
Correspondence Address:
Xiaofang Sun
Guangzhou Institute of Obstetrics and Gynecology, Key Laboratory for Major Obstetric Diseases of Guangdong Province, Key Laboratory of Reproduction and Genetics of Guangdong Higher Education Institutes, The Third Affiliated Hospital of Guangzhou Medical University, Guangzhou 510150, Guangdong
China
| How to cite this article: Wu T, Banerjee S, Deng J, Wu J, Huang H, Zheng H, Pan H, Wang Y, Peng Z, Sun X. Familial pityriasis rubra pilaris in a Chinese family caused by a novel mutation in CARD14 gene. Indian J Dermatol Venereol Leprol 2020;86:81-84 |
Sir,
Pityriasis rubra pilaris (PRP) (MIM# 173200) is a rare genodermatoses characterized by palmoplantar keratoderma and follicular hyperkeratotic papules. Generally, several follicular hyperkeratotic papules coalesce to form large, well-demarcated and scaly erythematous plaques interspersed by areas of normal skin.[1],[2] Erythroderma may result in severe cases. Familial PRP is inherited in an autosomal dominant manner. In addition, the clinical symptoms of familial PRP usually start to appear at the time of birth or in the first few years of life and patients gradually develop diffuse palmoplantar keratoderma, prominent follicular hyperkeratosis, and erythema.[3]
In this study, we investigated a Chinese family with familial PRP in the Department of Dermatology, Dermatology Hospital, Southern Medical University, Guangzhou, China. All the family members participating in this study had given their informed consent. The ethics committee of Dermatology Hospital, Southern Medical University had approved the study in accordance with the Declaration of Helsinki.
The proband was a 3-year-old Chinese girl who was born healthy [Figure - 1]a, but began to develop palmoplantar keratoderma along with erythema at 2 years of age. This was followed by development of large patches of hyperkeratosis and erythema with waxy keratotic plaques and fine scales on the knees, ankles, soles and gluteal cleft [Figure - 1]b, [Figure - 1]c, [Figure - 1]d, [Figure - 1]e, [Figure - 1]f, [Figure - 1]g. The surface of the keratotic plaques was covered with thin, hard, and rough scales. Biopsy from the right palm of the proband showed hyperkeratosis, acanthosis, and sparse lymphocytic infiltration in the superficial dermis [Figure - 1]h and i].
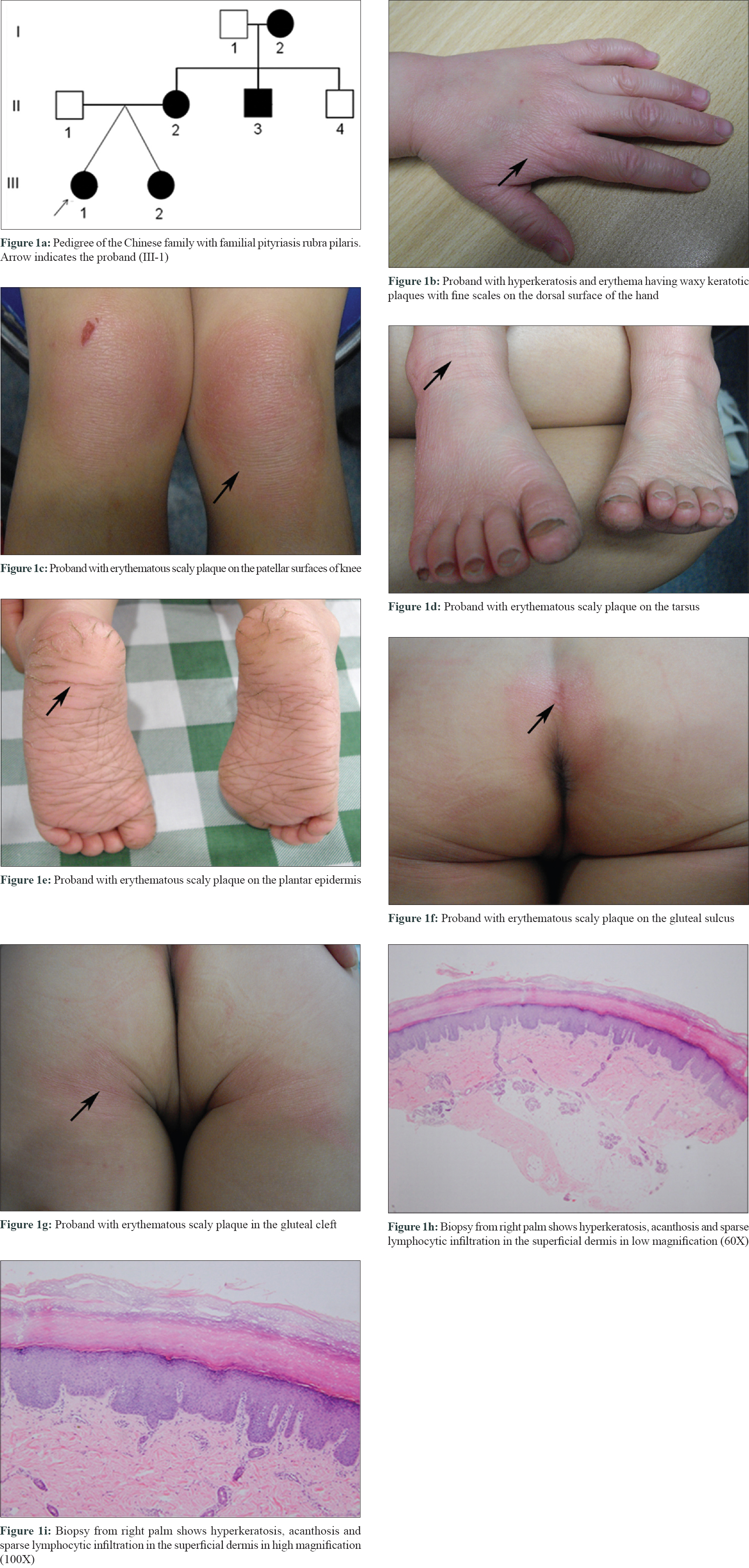 |
| Figure 1: |
Proband's mother presented with palmar hyperkeratosis and erythema during her childhood but milder than the proband, and the rashes gradually eased with increasing age. Proband's nonidentical twin sister (III-2) had erythema and hyperkeratosis on palms and soles epidermis only, similar to but milder than in the proband [Figure - 2]a, [Figure - 2]b, [Figure - 2]c. Proband's father was normal. Proband's maternal grandmother (I-2) and uncle (II-3) showed mild diffuse PPK without erythema.
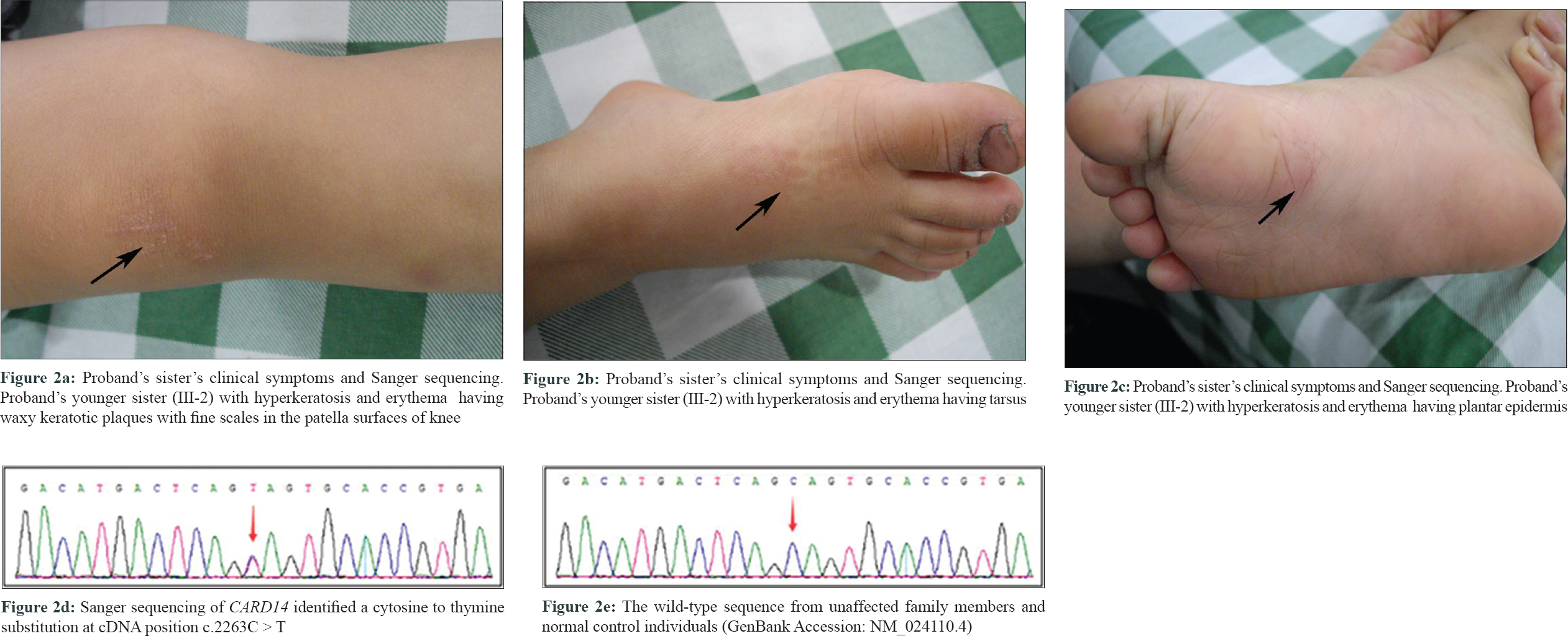 |
| Figure 2: |
General physical examination, systemic examination and developmental and cognitive assessment of the proband were normal. Dermatologic examination for dermatophytes and candida was negative from the hyperkeratotic skin of hands and feet of the proband. The rest of the skin and other ectodermal tissues were completely normal.
The patient and affected relatives were diagnosed with PRP according to the diagnostic criteria by two independent dermatologists.[4] To identify the genetic basis of the disease in this family, targeted next-generation sequencing and Sanger sequencing were performed. Targeted next-generation sequencing and Sanger sequencing identified a novel heterozygous mutation: c.2263C>T, p.Q755* in exon 16 of CARD14 in the proband (III: 1) and in all the affected family members. This mutation was absent in the unaffected family members and in normal controls [Figure - 2]d and [Figure - 2]e. This mutation co-segregated well with the disease phenotypes among the affected members in this family.
Here, we identified the first nonsense mutation in the human CARD14 gene, which results in the formation of truncated CARD14 protein in this three-generation Chinese family with familial PRP. This nonsense mutation leads to complete loss of the GuK domain from the CARD14 protein. This mutation is a loss-of function mutation according to the variants interpretation guidelines of the American College of Medical Genetics and Genomics.[5]
Our studied mutation is the first loss-of-function mutation which results in complete loss of the GuK domain of CARD14 protein. Familial PRP is an autosomal dominant genodermatoses, so it is quite expected that all the affected family members showed a similar disease phenotype as they are harboring the same pathogenic mutation in a heterozygous state.[3] However, in this family, we found a spectrum of disease phenotype categorized as severe, moderate, and mild. The proband had severe phenotype, while proband's mother and nonidentical twin sister had moderate phenotype, and proband's maternal grandmother and uncle had mild phenotype, although all of them were harboring the same heterozygous nonsense mutation in CARD14 gene. This can be explained by reduced penetrance and variable expressivity.[3] In this family, the extreme interindividual phenotypic diversity in the disease symptoms might be caused by the differences in the genetic background and the presence of “modifier genes” in each of the individuals.[2],[3]
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patients have given their consent for their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Acknowledgment
The authors would like to express their gratitude to the patient and her family members for participating in this study.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
| 1. |
Eytan O, Qiaoli L, Nousbeck J, van Steensel MA, Burger B, Hohl D, et al. Increased epidermal expression and absence of mutations in CARD14 in a series of patients with sporadic pityriasis rubra pilaris. Br J Dermatol 2014;170:1196-8.
[Google Scholar]
|
| 2. |
Fuchs-Telem D, Sarig O, van Steensel MA, Isakov O, Israeli S, Nousbeck J, et al. Familial pityriasis rubra pilaris is caused by mutations in CARD14. Am J Hum Genet 2012;91:163-70.
[Google Scholar]
|
| 3. |
Thomson MA, Moss C. Pityriasis rubra pilaris in a mother and two daughters. Br J Dermatol 2007;157:202-4.
[Google Scholar]
|
| 4. |
Griffiths WA. Pityriasis rubra pilaris. Clin Exp Dermatol 1980;5:105-12.
[Google Scholar]
|
| 5. |
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405-24.
[Google Scholar]
|
Fulltext Views
4,131
PDF downloads
1,828





![[Figure - 1]](#fig_ijdvl_2020_86_1_81_271491_f1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2020_86_1_81_271491_f2.jpg){kind=link}