Translate this page into:
H syndrome with histological features of Langerhans cell histiocytosis
2 Department of Medical Genetics, Anatomy and Cytology, Farhat Hached Hospital, Sousse, Tunisia
3 Department of Biochemistry and Molecular Biology, Trousseau Hospital, Paris, France
4 Department of Pathological Anatomy and Cytology, Anatomy and Cytology, Farhat Hached Hospital, Sousse, Tunisia
Correspondence Address:
Mouna Korbi
Department of Dermatology, Farhat Hached Hospital, Sousse
Tunisia
| How to cite this article: Korbi M, Aounallah A, Hmida D, Ghariani N, Saidi W, Boussofara L, Belajouza C, Jonard L, Sriha B, Saad A, Denguezli M, Nouira R. H syndrome with histological features of Langerhans cell histiocytosis. Indian J Dermatol Venereol Leprol 2016;82:702-704 |
Sir,
The H syndrome (OMIM 602782) is a very rare autosomal recessive disorder first described by Molho-Pessach in 2008 and refers to a constellation of clinical and laboratory findings.[1],[2],[3],[4] This disorder has been recently shown to be caused by mutations in the SLC29A3 gene encoding the human equilibrative nucleoside transporter.[5] We report a novel case of H syndrome with a particular histological and genetic presentation.
The patient followed up with us since the age of 14 years for progressive sclerodermatous thickening of the skin, with overlying hyperpigmentation and hypertrichosis on the limbs [Figure - 1]. In addition, he was followed up in the endocrinology department since the age of 16 years for insulin-dependent diabetes mellitus, short stature and dysmorphic features. The child also had sensorineural hearing loss from the age of 9 years. He was born after a normal pregnancy to non-related parents of Tunisian origin. His brother and two sisters were healthy. On physical examination, we noted bilateral gynecomastia [Figure - 1], short stature, hepatosplenomegaly, flexion contractures of the fingers and toes, hallux valgus and a micropenis. Indurated and hyperpigmented patches were present on the middle and lower half of the body, extending over the trunk and neck. He was not found to have cardiovascular or ocular anomalies. Laboratory investigations revealed low rates of insulin growth factor type 1, insulin growth factor binding protein type 3 and testosterone. A growth hormone stimulation test with arginine did not indicate growth hormone deficiency. Thyroid function tests and karyotype were normal.
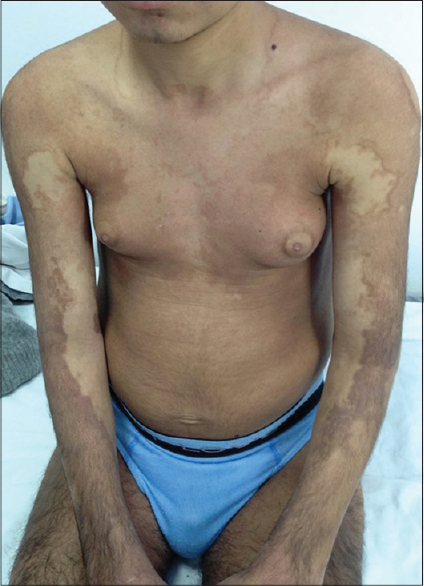 |
| Figure 1: Extensive hyperpigmentation over upper limbs and thighs, and prominent gynecomastia |
Histopathological examination of involved skin showed hyperpigmentation of the basal layer with infiltration by histiocytes [Figure - 2]. Immunohistochemistry showed positivity with both CD1a and PS100 [Figure - 3]. However, CD68 was negative. A perivascular mononuclear infiltrate with plasma cells and mast cells throughout the dermis and subcutaneous fat was noted. Blood samples were collected from the patient and his parents. Informed consent was obtained from each of them. Mutation analysis of genomic DNA confirmed the diagnosis of H syndrome in our patient and showed that he was a compound heterozygote for two types of mutations in exon 6 of the SLC29A3 gene (described below).
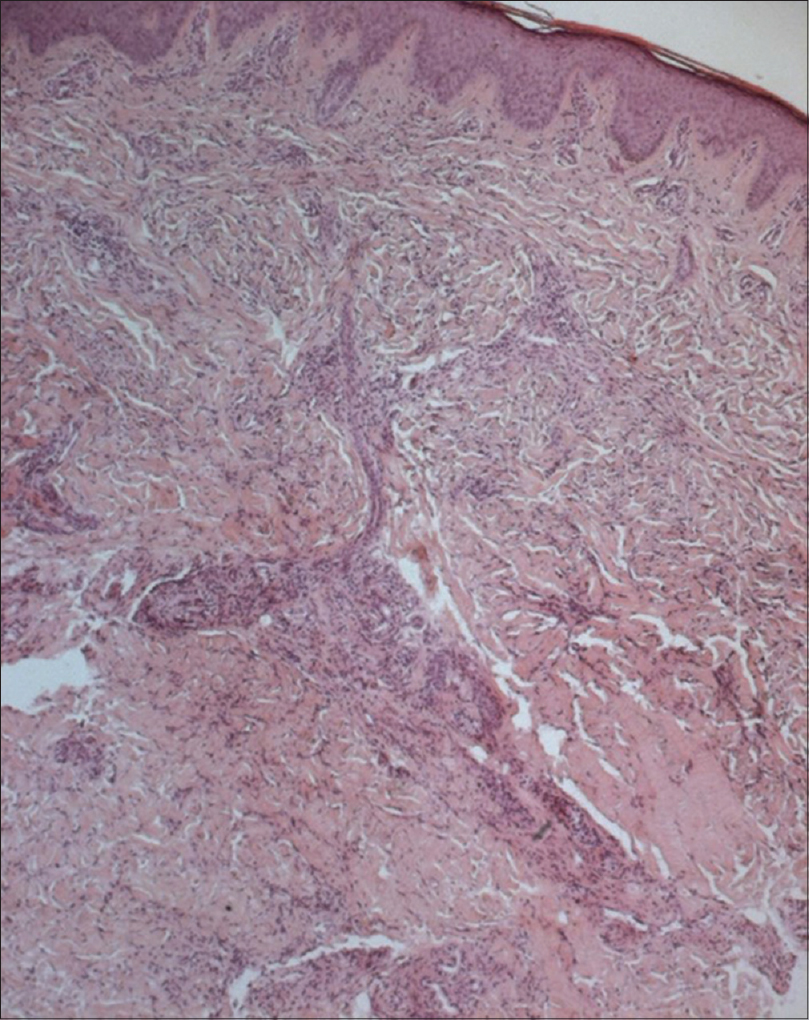 |
| Figure 2: Marked fi brosis of the dermis and superfi cial subcutis with chronic perivascular infl ammatory infi ltrates (H and E, ×400) |
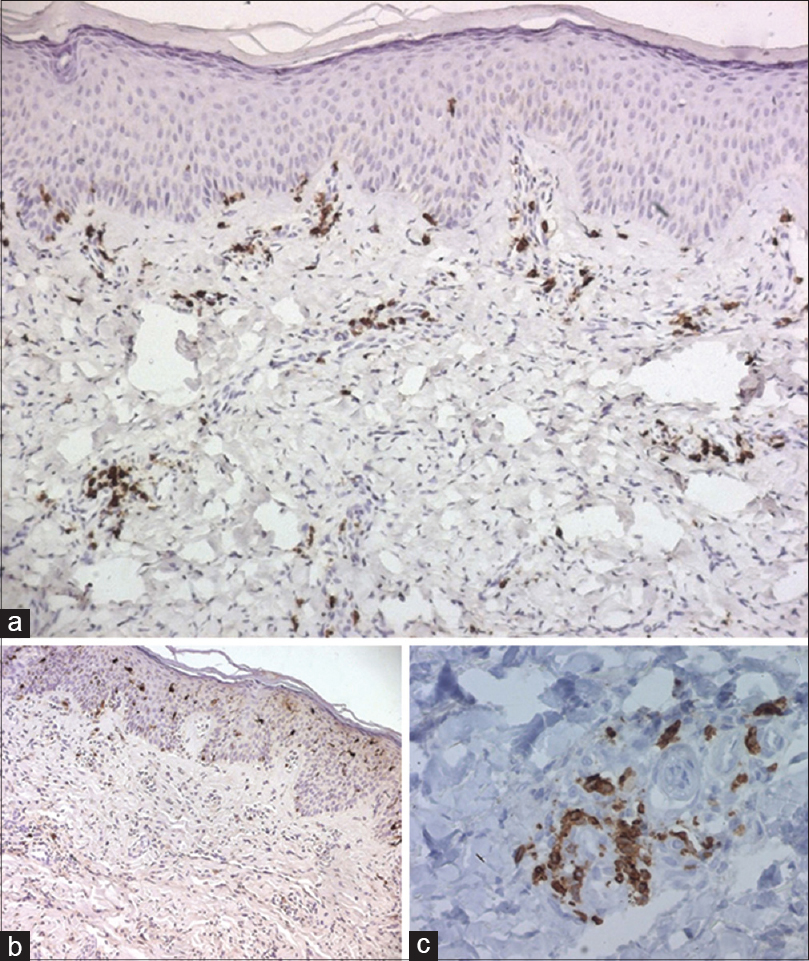 |
| Figure 3: (a) The lymphoid cells are seen to be mostly T cells (CD3+) (b) with histiocytes which are CD1a+ (c) histiocytes are also positive with PS100 |
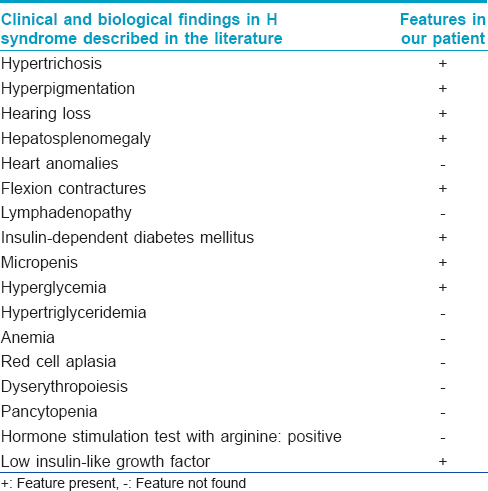
More than a hundred cases of the H syndrome have been reported.[2] Major systemic features include hearing loss, heart anomalies, hepatosplenomegaly, hypogonadism and hyperglycemia. A hallmark of this syndrome is the cutaneous hyperpigmentation which is accompanied by sclerodermatous thickening and hypertrichosis.[3] The lower limbs are the most common site described with cutaneous hyperpigmentation. Flexion contractures of the fingers and toes are the second most common feature.[3] In a recent study which focuses on the spectrum of radiological findings in the H syndrome, severe symmetrical deformation was demonstrated.[6] Sensorineural hearing loss is the third most common clinical finding. A recent report describes two cases of H syndrome who had hearing loss as a prominent feature.[7] Lymphadenopathy has been described as another feature.[3] According to a recent review, insulin-dependent diabetes mellitus was observed in 18 (23%) patients, and this may be the sole presentation.[8] Micropenis has also been observed.[3],[4] Further, several other biological anomalies such as hypertriglyceridemia, anemia, red cell aplasia/dyserythropoiesis and pancytopenia have been described in the literature.[3],[4],[9] Hormonal evaluation was done in some studies. A growth hormone stimulation test with arginine was consistent with growth hormone deficiency, providing an explanation for the short stature.[1],[4] Low insulin-like growth factor 1 level was reported as another contributing factor to short stature. The sex hormone profile showed relatively high gonadotropins and low testosterone levels, indicating primary hypogonadism.[1],[4] Our patient had many of these findings [Table - 1].
Skin biopsy specimens in the H syndrome demonstrate polyclonal perivascular lympho-histiocytic infiltrates and hyperpigmentation of the basal layer,[5] as with our patient. However, in our case, immunohistochemical stains showed histiocytes positive for CD1a and PS100, a feature not described earlier. Our patient could therefore be considered as having a variant of H syndrome, within the spectrum of inherited histiocytosis.
Till now, only twenty mutations have been identified in the H syndrome.[3] Our patient had two different mutations previously described: a missense mutation c. 1088G>A changing arginine 363 to glutamine (p. Arg363Gln) previously detected in Arab patients and a splice site mutation, c. 300+1G>A which was reported in Pakistani patients.[5] The spectrum of phenotypes related to SLC29A3 mutations appears to be very wide, ranging from a severe syndrome as in our patient, to a very mild clinical presentation. No clear correlation between any of the clinical features and a specific mutation has been found. In fact, patients with a mild phenotype have been identified after mutation screening in families with severely affected individuals.[4] There is no specific treatment for this syndrome, and genetic counseling is essential.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
| 1. |
Molho-Pessach V, Agha Z, Aamar S, Glaser B, Doviner V, Hiller N, et al. The H syndrome: A genodermatosis characterized by indurated, hyperpigmented, and hypertrichotic skin with systemic manifestations. J Am Acad Dermatol 2008;59:79-85.
[Google Scholar]
|
| 2. |
Sharad M, Vaishali M, Asit M, Achok KK, Lalit GK. The H syndrome. Indian J Peadiatric Dermatol 2015;16:102-4.
[Google Scholar]
|
| 3. |
Molho-Pessach V, Ramot Y, Camille F, Doviner V, Babay S, Luis SJ, et al. Hsyndrome: The first 79 patients. J Am Acad Dermatol 2014;70:80-8.
[Google Scholar]
|
| 4. |
Molho-Pessach V, Lerer I, Abeliovich D, Agha Z, Abu Libdeh A, Broshtilova V, et al. The H syndrome is caused by mutations in the nucleoside transporter hENT3. Am J Hum Genet 2008;83:529-34.
[Google Scholar]
|
| 5. |
Jonard L, Couloigner V, Pierrot S, Louha M, Gherbi S, Denoyelle F, et al. Progressive hearing loss associated with a unique cervical node due to a homozygous SLC29A3 mutation: A very mild phenotype. Eur J Med Genet 2012;55:56-8.
[Google Scholar]
|
| 6. |
Hiller N, Zlotogorski A, Simanovsky N, Ingber A, Ramot Y, Molho-Pessach V. The spectrum of radiological findings in H syndrome. Clin Imaging 2013;37:313-9.
[Google Scholar]
|
| 7. |
Ramot Y, Sayama K, Sheffer R, Doviner V, Hiller N, Kaufmann-Yehezkely M, et al. Early-onset sensorineural hearing loss is a prominent feature of H syndrome. Int J Pediatr Otorhinolaryngol 2010;74:825-7.
[Google Scholar]
|
| 8. |
Broshtilova V, Ramot Y, Molho-Pessach V, Zlotogorski A. Diabetes mellitus may be the earliest and sole manifestation of the H syndrome. Diabet Med 2009;26:1179-80.
[Google Scholar]
|
| 9. |
Priya TP, Philip N, Molho-Pessach V, Busa T, Dalal A, Zlotogorski A. H syndrome: Novel and recurrent mutations in SLC29A3. Br J Dermatol 2010;162:1132-4.
[Google Scholar]
|
Fulltext Views
3,933
PDF downloads
2,743





![[Figure - 1]](#fig_ijdvl_2016_82_6_702_185044_f1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2016_82_6_702_185044_f2.jpg){kind=link}
![[Figure - 3]](#fig_ijdvl_2016_82_6_702_185044_f3.jpg){kind=link}
![[Table - 1]](#tbl_ijdvl_2016_82_6_702_185044_t4.jpg){kind=link}