Translate this page into:
Hemophagocytic lymphohistiocytosis: A rare, potentially fatal complication in subcutaneous panniculitis like T cell lymphoma
2 Department of Pathology, All India Institute of Medical Sciences, Bhubaneswar, Odisha, India
3 Department of Radiotherapy, All India Institute of Medical Sciences, Bhubaneswar, Odisha, India
Correspondence Address:
Chandra Sekhar Sirka
Department of Dermatology, All India Institute of Medical Sciences, Bhubaneswar, Odisha
India
| How to cite this article: Sirka CS, Pradhan S, Patra S, Padhi S, DasMajumdar SK, Panda D. Hemophagocytic lymphohistiocytosis: A rare, potentially fatal complication in subcutaneous panniculitis like T cell lymphoma. Indian J Dermatol Venereol Leprol 2019;85:481-485 |
Abstract
Subcutaneous panniculitis-like T cell lymphoma is a rare subtype of cutaneous lymphomas with distinct clinical, histological and immunophenotypic characteristics, as well as an indolent clinical course. Rarely, it may be complicated with hemophagocytic lymphohistiocytosis: a hyperinflammatory syndrome which, if not diagnosed early, carries a dismal outcome. In this article, we describe a case of subcutaneous panniculitis-like T cell lymphoma in a middle-aged female patient which was complicated with secondary hemophagocytic lymphohistiocytosis with a favorable outcome following etoposide-based therapy. The various histological mimics of subcutaneous panniculitis-like T cell lymphoma and the management options are also briefly discussed.
Introduction
Subcutaneous panniculitis-like T cell lymphoma is an uncommon type of skin lymphoma (<1% of all cutaneous lymphomas) characterized by plaques and subcutaneous nodules with ulceration. Histologically, this is characterized by subcutaneous fat infiltration with atypical T lymphocytes rimming mature adipocytes, mimicking panniculitis.[1] Subcutaneous panniculitis-like T cell lymphoma can be complicated with hemophagocytic lymphohistiocytosis – a rare, potentially fatal, hyperinflammatory syndrome characterized by severe constitutional symptoms, splenomegaly and other deranged laboratory parameters, which if not diagnosed early carries a poor prognosis.[2] In this article, we aim to present a case of subcutaneous panniculitis-like T cell lymphoma complicated with hemophagocytic lymphohistiocytosis which was treated with etoposide-based chemotherapy.
Case Report
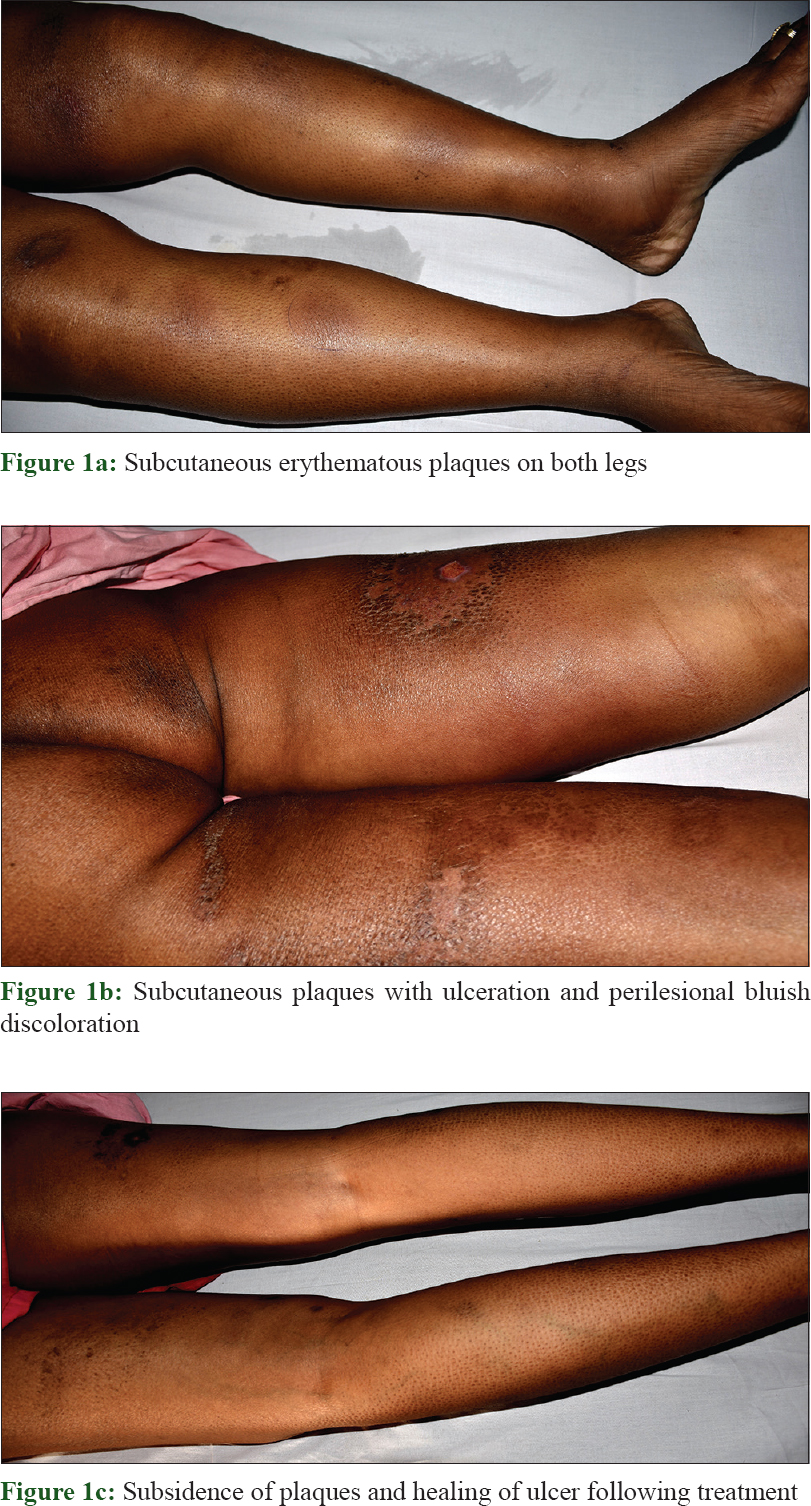
A 38-year-old female patient presented to the dermatology outpatient department of All India Institute of Medical Sciences, Bhubaneswar with high-grade fever, night sweating, and multiple, painful reddish nodules over the trunk and extremities for 6 months. On examination, there were multiple, tender, erythematous subcutaneous plaques and nodules with massive induration varying in size from 2 to 15 cm. A few plaques and nodules had mild scaling (following subsidence of erythema) and some were ulcerated with perilesional bluish pigmentation. The older lesions were subsiding leaving bluish discoloration [Figure - 1]a and [Figure - 1]b. General examination revealed high-grade fever (106°F), conjunctival pallor; other systemic examinations were unremarkable except hepatosplenomegaly.
 |
| Figure 1 |
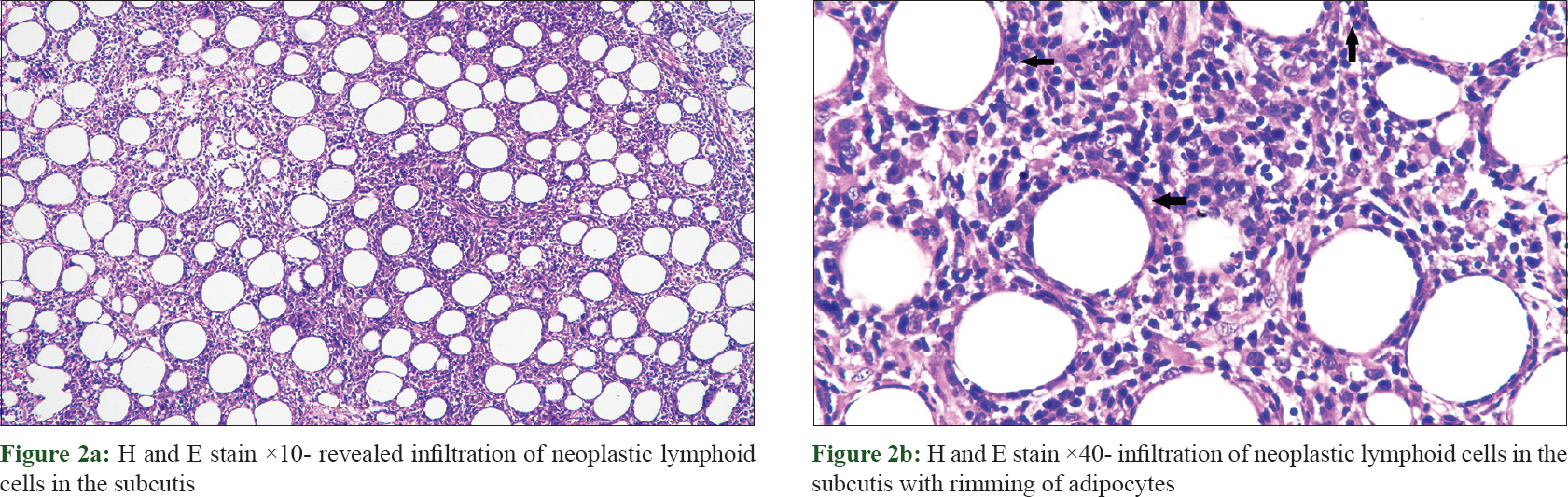
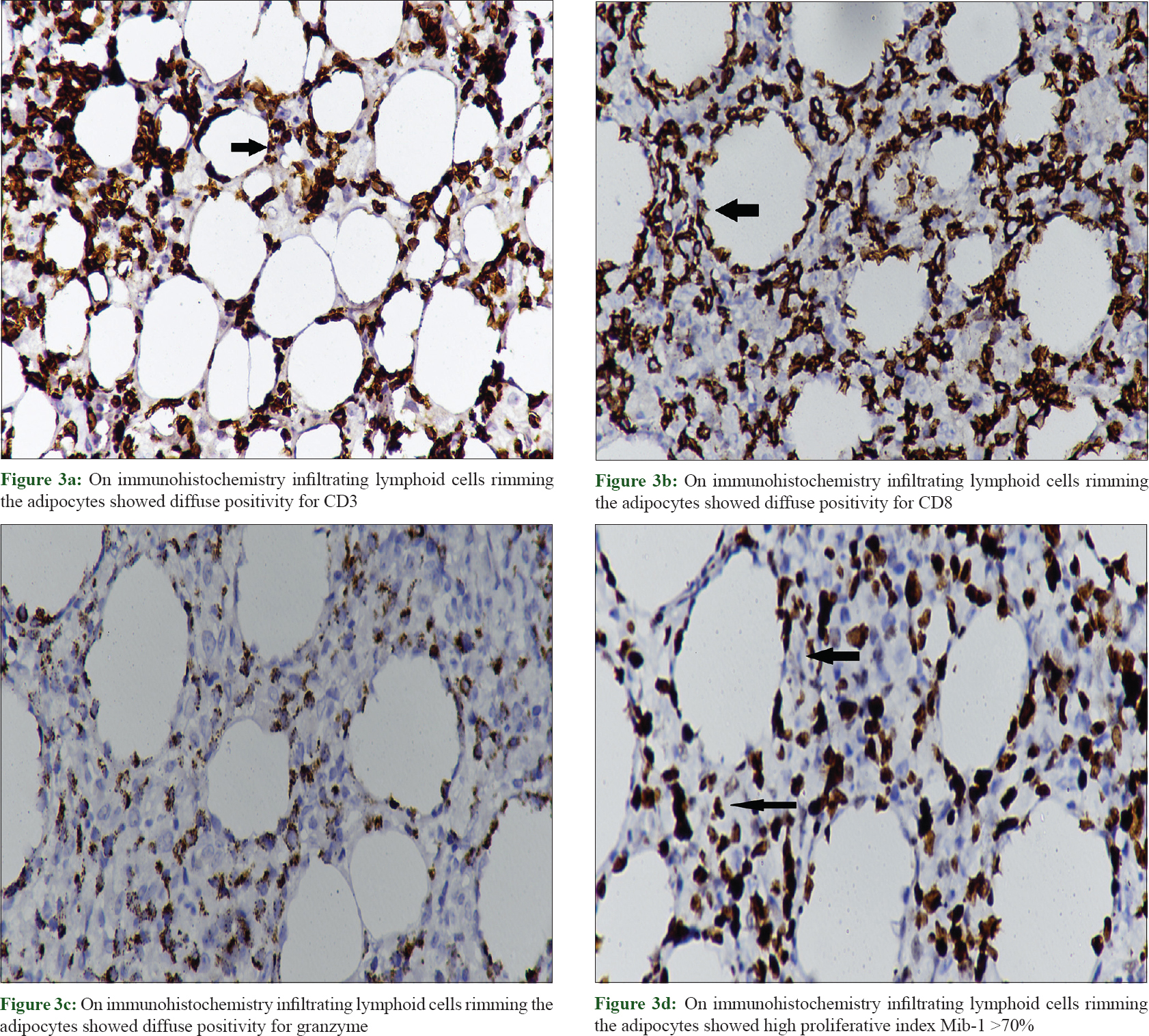
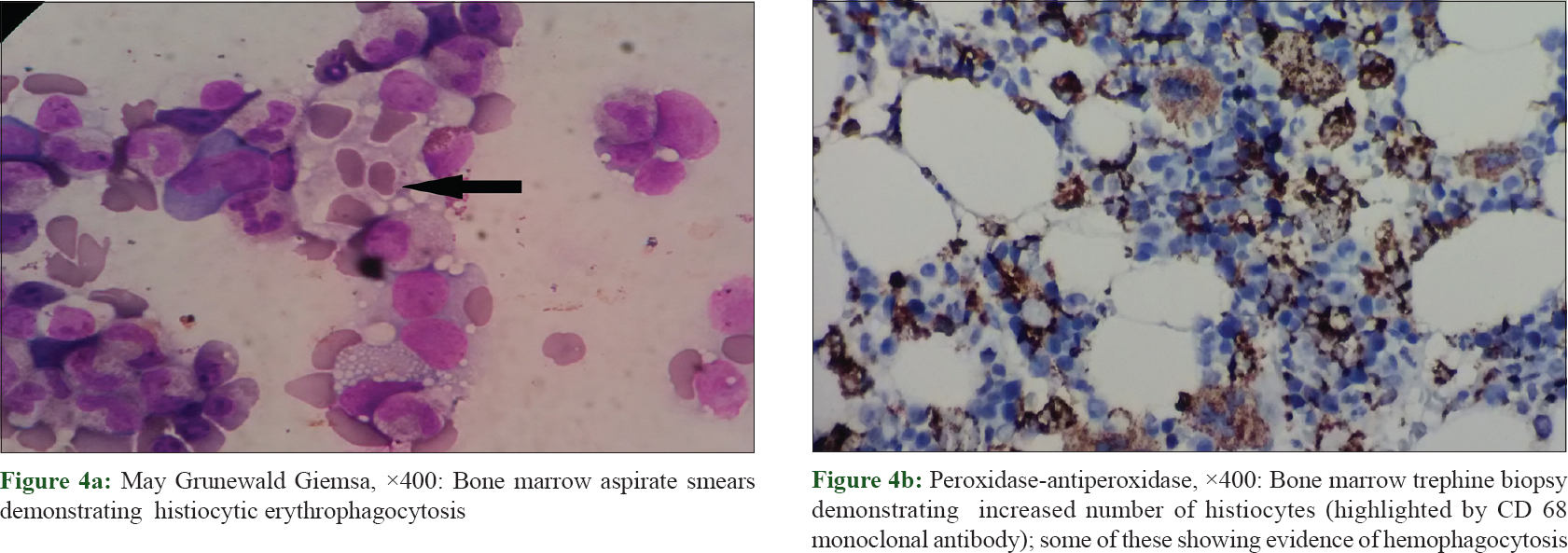
Routine laboratory investigations revealed microcytic hypochromic anemia [hemoglobin, 74 g/L (ref. 120–140 g/L); hematocrit, 29% (ref. 36–46%) and mean corpuscular volume, 67 fL (ref. 80–98 fL)], total leukocyte count, 7.5 × 10[9]/L (ref. 4–11 × 10[9]/L), total platelet count, 90 × 10[9]/L (ref. 150–450 10[9]/L), elevated liver transaminases (twenty times from the normal upper limit) and bilirubin (total, 5.7 mg/ dl, direct, 4.3 mg/ dl). Her autoimmune work-up was negative for serum antinuclear antibody and anti-double stranded DNA. Thoracic and abdominal contrast-enhanced computerized tomogram scan showed hepatosplenomegaly but no lymphadenopathy. Histopathological evaluation of the biopsy from the plaque on the right thigh revealed infiltration of atypical lymphoid cells with irregular nuclear contour in the subcutis along with rimming of adipocytes [Figure - 2]a and [Figure - 2]b. There were small foci of fat necrosis with karyorrhectic debris and collection of macrophages. There was no evidence of epidermotropism or infiltration of upper dermis. On immunohistochemistry, the lymphoid cells showed strong and diffuse positivity for CD3, CD8 and granzyme with a high proliferative index (Mib-1 >70%) [Figure - 3]a, [Figure - 3]b, [Figure - 3]c, [Figure - 3]d. Other markers such as CD20, CD4 and CD56 were negative. Based upon the histomorphological and immunohistochemistry findings, a diagnosis of subcutaneous panniculitis-like T cell lymphoma was made. The patient was started on oral prednisolone 60 mg. Even after 4 weeks of treatment, systemic symptoms such as high-grade fever and sweating were unremitting along with persistence of other factors such as anemia, thrombocytopenia, deranged liver function test and hepatosplenomegaly, leading to a suspicion of hemophagocytic lymphohistiocytosis. Further laboratory investigation showed hyperferritinemia (6160 ng/ml; reference range, <500 ng/ml), fasting hypertriglyceridemia (379 mg/dl) and hypofibrinogenemia (0.9 g/L). Bone marrow aspiration and trephine biopsy revealed histiocytic erythrophagocytosis [Figure - 4]a and [Figure - 4]b. The patient satisfied six of eight HLH-2004 diagnostic criteria; and the probability score (HScore) for a diagnosis hemophagocytic lymphohistiocytosis was 273 (cut-off score, 169; sensitivity, 93%; specificity, 86%; accurate classification of hemophagocytic lymphohistiocytosis in 90% of the patients).[3],[4] The patient was started on etoposide and dexamethasone; and after 3 weeks, systemic symptoms improved along with decrease in skin lesions [Figure - 1]c. At present, the patient is under treatment with cyclophosphamide, doxorubicin, vincristine and prednisolone chemotherapy.
 |
| Figure 2 |
 |
| Figure 3 |
 |
| Figure 4 |
Discussion
There is a wide range of conditions which can have plaques and nodules with ulceration such as lupus panniculitis, cytophagic histiocytic panniculitis, Subcutaneous panniculitis-like T-cell lymphoma, pancreatic panniculitis, nodular vasculitis and erythem nodosum leprosum. Histopathological evaluation of the representative biopsy can differentiate majority of the conditions.[5]
The index patient satisfied 5 out of the 6 tested hemophagocytic lymphohistiocytosis 2004 criteria (fever >38.5°C, splenomegaly, peripheral blood cytopenias, hypertriglyceridemia and hypofibrinogenemia, hemophagocytosis in the bone marrow and hyperferritinemia).[3] Recently, a composite probability score (HScore) has been proposed by Fardet et al. for an early diagnosis of hemophagocytic lymphohistiocytosis.[4] This score takes into consideration several clinic-laboratory parameters such as the presence of immunosuppression, fever, cytopenia (s), splenohepatomegaly, hyperferritinemia, hypertriglyceridemia, evidence of histiocytic hemophagocytosis in bone marrow aspirate and elevated liver transaminases. A composite score of 169 had a sensitivity of 93% and specificity of 86%, with accurate classification of hemophagocytic lymphohistiocytosis in 90% of the patients.[4] Our patient had an H score of 273 which led to a definite diagnosis of hemophagocytic lymphohistiocytosis. Bone marrow evaluation did not show any evidence of lymphomatous infiltrate.
According to the new World Health Organization (WHO) European Organization for Research and Treatment of Cancer (EORTC) classification, lymphomas expressing T cell receptor–alpha/beta are normally CD8 positive and CD56 negative, are usually restricted to the subcutaneous tissue and carry a favourable prognosis. In contrast, cases with CD4 and CD8-negative and CD56-positive phenotype usually express T-cell receptor–gamma/delta. The latter may involve the epidermis with a poor prognosis, and are placed in a new category of primary cutaneous peripheral T cell lymphomas—cutaneous gamma/delta T-cell lymphoma.[1],[6],[7]
In 2008, the EORTC Cutaneous Lymphoma Group first reported the clinicopathological, immunophenotypic and follow-up data of 83 cases of subcutaneous panniculitis-like T cell lymphoma.[1] Sixty-three cases were categorized as alpha/beta phenotype [21 males, 42 females, median age; 36 years (12/63; younger than 20 years)] and the remaining 20 were that of gamma/delta phenotype (7 males, 13 females, median age; 59 years). Eleven cases (17%) with alpha/beta phenotype were complicated with hemophagocytic lymphohistiocytosis; all had evidence of histiocytic hemophagocytosis in the bone marrow; 7 of the 11 cases had a fatal outcome. On the other hand, 9 of 20 cases in the latter subgroup had associated hemophagocytic lymphohistiocytosis and 7 had a fatal outcome. Subcutaneous panniculitis-like T cell lymphoma–alpha/beta patients without hemophagocytic lymphohistiocytosis had a significantly better survival than patients with hemophagocytic lymphohistiocytosis (5-year overall survival: 91% vs 46%; P <.001). While the alpha/beta group had a favorable prognosis (5-year overall survival: 82%), gamma/delta group often showed epidermal involvement and/or ulceration and a poor prognosis (5-year overall survival: 11%), irrespective of the presence of hemophagocytic lymphohistiocytosis or type of treatment.[1]
Systemic corticosteroids have been considered excellent first-line agents in various reports and studies.[8],[9],[10] Other treatment options such as combination chemotherapy with cyclophosphamide, doxorubicin, vincristine and prednisolone chemotherapy regimen and cyclosporine alone have been tried successfully in various cases.[11] In our case, the patient was diagnosed as subcutaneous panniculitis-like T cell lymphoma–alpha/beta subtype based upon the clinical features, histopathology and immunohistochemistry. Even though skin lesions improved with 4-week oral prednisolone therapy, systemic symptoms such as continued pyrexia, sweating, deranged liver function tests, hepatosplenomegaly and cytopenias persisted, which led to further evaluation for secondary hemophagocytic lymphohistiocytosis. The patient was then started with etoposide combined with steroid-based regimen, following which the fever subsided and liver function tests started improving. The patient is currently continuing her treatment without any fever, skin lesions and constitutional symptoms.
To conclude: hemophagocytic lymphohistiocytosis is a potentially fatal condition which can rarely complicate indolent T-cell lymphomas like subcutaneous panniculitis-like T cell lymphoma. Therefore, patients with subcutaneous panniculitis-like T cell lymphoma with severe constitutional symptoms should be thoroughly evaluated for associated hemophagocytic lymphohistiocytosis. Early usage of immunosuppressive and/or etoposide-based therapy might be useful in reducing the severity and mortality.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form, the patient has given his consent for her images and other clinical information to be reported in the journal. The patient understand that name and initials will not be published and due efforts will be made to conceal identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
| 1. |
Willemze R, Jansen PM, Cerroni L, Berti E, Santucci M, Assaf C, et al. Subcutaneous panniculitis-like T-cell lymphoma: Definition, classification, and prognostic factors: An EORTC Cutaneous Lymphoma Group Study of 83 cases. Blood 2008;111:838-45.
[Google Scholar]
|
| 2. |
Padhi S, Varghese RG, Ramdas A, Phansalkar MD, Sarangi R. Hemophagocytic lymphohistiocytosis: Critical reappraisal of a potentially under-recognized condition. Front Med 2013;7:492-8.
[Google Scholar]
|
| 3. |
Henter JI, Horne A, Aricó M, Egeler RM, Filipovich AH, Imashuku S, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer 2007;48:124-31.
[Google Scholar]
|
| 4. |
Fardet L, Galicier L, Lambotte O, Marzac C, Aumont C, Chahwan D, et al. Development and validation of the HScore, a score for the diagnosis of reactive hemophagocytic syndrome. Arthritis Rheumatol 2014;66:2613-20.
[Google Scholar]
|
| 5. |
Bosisio F, Boi S, Caputo V, Chiarelli C, Oliver F, Ricci R, et al. Lobular panniculitic infiltrates with overlapping histopathologic features of Lupus panniculitis (Lupus profundus) and subcutaneous T-cell lymphoma: A conceptual and practical dilemma. Am J Surg Pathol 2015;39:206-11.
[Google Scholar]
|
| 6. |
Slater DN. The new World Health Organization-European Organization for Research and Treatment of Cancer classification for cutaneous lymphomas: A practical marriage of two giants. Br J Dermatol 2005;153:874-80.
[Google Scholar]
|
| 7. |
Singh A, Kumar J, Kapur S, Ramesh V. Subcutaneous panniculitis-like T-cell cutaneous lymphoma. Indian J Dermatol Venereol Leprol 2008;74:151-3.
[Google Scholar]
|
| 8. |
Guenova E, Schanz S, Hoetzenecker W, DeSimone JA, Mehra T, Voykov B, et al. Systemic corticosteroids for subcutaneous panniculitis-like T-cell lymphoma. Br J Dermatol 2014;171:891-4.
[Google Scholar]
|
| 9. |
Hoque SR, Child FJ, Whittaker SJ, Ferreira S, Orchard G, Jenner K, et al. Subcutaneous panniculitis-like T-cell lymphoma: A clinicopathological, immunophenotypic and molecular analysis of six patients. Br J Dermatol 2003;148:516-25.
[Google Scholar]
|
| 10. |
Hu ZL, Sang H, Deng L, Li Z. Subcutaneous panniculitis-like T-cell lymphoma in children: A review of the literature. Pediatr Dermatol 2015;32:526-32.
[Google Scholar]
|
| 11. |
Lee WS, Hwang JH, Kim MJ, Go SI, Lee A, Song HN, et al. Cyclosporine A as a primary treatment for panniculitis-like T cell lymphoma: A case with a long-term remission. Cancer Res Treat 2014;46:312-6.
[Google Scholar]
|
Fulltext Views
3,777
PDF downloads
2,761





![[Figure - 1]](#fig_ijdvl_2019_85_5_481_242674_f1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2019_85_5_481_242674_f2.jpg){kind=link}
![[Figure - 3]](#fig_ijdvl_2019_85_5_481_242674_f3.jpg){kind=link}
![[Figure - 4]](#fig_ijdvl_2019_85_5_481_242674_f4.jpg){kind=link}