Translate this page into:
Hutchinson-Gilford progeria syndrome
2 Department of Pediatrics, SMS Medical College, Jaipur, India
Correspondence Address:
Uma Shankar Agarwal
397, Shankar Niwas, Shri Gopal Nagar, Near Gujjar Ki Thaddi, Gopalpura Bypass, Mansarovar, Jaipur
India
| How to cite this article: Agarwal US, Sitaraman S, Mehta S, Panse G. Hutchinson-Gilford progeria syndrome. Indian J Dermatol Venereol Leprol 2010;76:591 |
Abstract
Progeria is a rare genetic disorder characterized by premature aging, involving the skin, bones, heart, and blood vessels. We report a 4-year-old boy who presented with clinical manifestations of progeria. He had characteristic facies, prominent eyes, scalp and leg veins, senile look, loss of scalp hair, eyebrows and eyelashes, stunted growth, and sclerodermatous changes. The present case is reported due to its rarity.Introduction
Hutchison-Gilford progeria syndrome (HGPS) was first reported in the literature in 1886 by Hutchinson as a case of "congenital absence of hair and its appendages" and Gilford introduced the term progeria. Although senile degeneration occurs, many features of aging like presbycusis, presbyopia, cataract, arcus senilis, and osteoarthritis are not seen. This syndrome may only mimic portions of the aging process.
The HGPS phenotype is so constant that patients with the condition bear resemblance to one another. It is a rare hereditary disor-der with approximately 100 cases reported in the literature, [1] which prompted us to report this case.
Case Report
A 4-year-old boy presented to the skin out patient department of our hospital with loss of scalp hair, eyebrows and eye lashes since 1 year of age along with stunted growth as a major complaint. On examination he had distinctive facies with prominent eyes and scalp veins, diffuse hair loss, small ear lobes, and beaked nose [Figure - 1]. He had protuberant abdomen with mottled pigmentation and sclerodermatous changes [Figure - 2]. He had a varus derformity of the knees with prominent superficial leg veins [Figure - 3],[Figure - 4]. His teeth were normal. The hands were short and stubby, with small and dystrophic nails [Figure - 5]. The patient′s genitals were normal. His voice was high-pitched and he had stunted growth. His height was 82 cm (corresponding height at 50th percentage in a normal 4-year-old boy is 102 cm) and weight was 8.5 Kg (corresponding weight at 50th percentage in a normal 4-year-old boy is 16 kg). The patient′s intelligence was normal. Rest of the physical examination was normal.
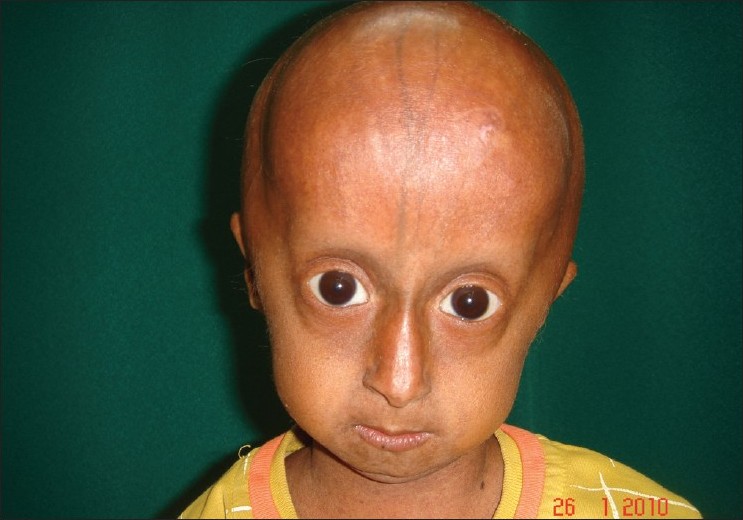 |
| Figure 1 : Face showing alopecia, loss of eyebrow and eyelashes, prominent eyes and tapered nasal tip |
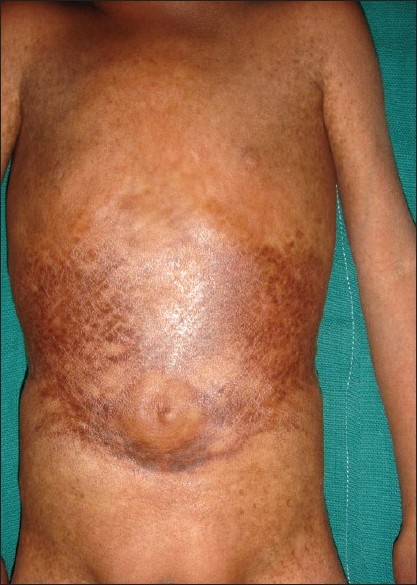 |
| Figure 2 : Mottled pigmentation and sclerodermatous changes on abdomen |
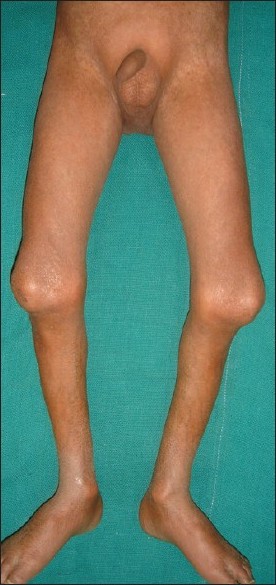 |
| Figure 3 : Knee joint deformity |
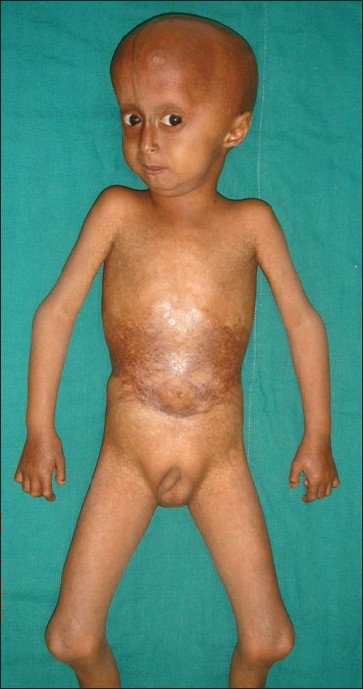 |
| Figure 4 : General appearance with short stature |
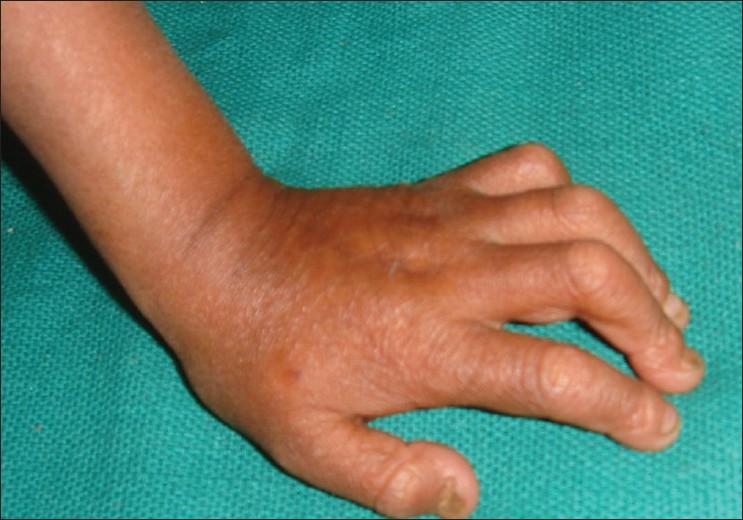 |
| Figure 5 : Short and stubby hands with small and dystrophic nails |
Complete hemogram, erythrocyte sedimentation rate, urine examination, and liver and renal function tests were within normal limit. The serum lipid profile showed a decrease in high density lipoprotein with the other parameters being normal. X-ray findings of extremities showed osteoporotic changes with calcification around knee joint, and flexion deformity of fingers of hands. Radiographic study of skull and chest was normal. Fundoscopy, ECG, and ultrasonography of abdomen did not show any anomaly. The patient could not afford to get a DEXA scan.
Discussion
Progeria is a premature aging syndrome. The earliest feature is growth deficiency in the first year of life. These patients exhibit characteristic facies, alopecia in first 2 years of life, short stature, sclerodermatous change, "sculputured nasal tip", prominent scalp veins, and joint deformities. [2] Exact etiology of progeria is not known, but increasing evidence supports the de novo point mutations in lamin A (LMNA) gene as the causative factor. Prelamin A is a protein located on the nuclear membrane of cells, it needs to be cleaved to form Lamin A, a process defective in patients of progeria. The increased prelamin A causes nuclear blebbing and altered shape of the nucleus. In HGPS, the sequencing of LMNA, located on chromosome 1q, revealed that 18 out of 20 classical cases of HGPS harbored an identical de novo single-base substitution (cytosine to thymine), within exon 11. [3] How the altered shape of the nucleus leads to the premature aging symptoms is however not known. There is evidence of increased production of hyaluronic acid by the fibroblast in these patients. [1] Other etiological possibilities include increased tropoelastin production and accumulation of type IV collagen by interaction between activated T lymphocyte and fibroblast. [4]
HGPS has an incidence of about 1 in 4 to 8 million newborns. It occurs due to a sporadic mutation and hence does not generally run in families. However, there have been very rare reports of progeria affecting more than one person in a family, including a report of five cases in a single family from India in 2005.
Our case had classical manifestations of progeria. Typical hair and face involvement excluded acrogeria. Werner′s syndrome usually presents between 14 and 18 years of age with short stature, immature sexual development, cataract, glaucoma, and sclerodermatous changes. Rothmund-Thomson syndrome appears between 3 and 6 months of age with poikilodermatous skin, photosensitivity, and cataract. Cockayne′s syndrome is manifested as facial erythema in butterfly distribution, photosensitivity, ocular defects, and a "micky mouse" appearance with disproportionately large hands and feet and protruding ears.
Cutaneous manifestations are earlier to appear followed by skeletal and cardiovascular systems. In our case cardiovascular system was normal. Skeletal abnormalities include osteolysis, osteoporosis, dystrophic clavicles, coxa valga, "horse riding" stance, thinning of cranial bones, delayed closure of cranial sutures and anterior fontanelle. [5]
Long-term follow-up is needed to observe the cardiovascular and skeletal abnormalities in these patients. Death usually occurs in second decade as a result of severe generalized atherosclerosis leading to cardiovascular or cerebrovascular complications. Other common complications include osteoporosis, pathological fractures, dental anomalies, and malnutrition. Low-dose aspirin is recommended as prophylaxis to prevent atherosclerotic changes. In vitro studies in mice suggest a possible role for the use of farnesyltransferase inhibitors (FTIs) in progeria. FTIs allow prelamin A to be correctly incorporated into the nuclear lamina, thus correcting the structural and functional nuclear defects. A phase II trial of lonafarnib, an FTI, in progeria is currently ongoing in USA, along with two other drugs: pravastatin, a cholesterol-lowering agent and zoledronic acid, a biphosphonate, to prevent atherosclerosis and osteoporosis, respectively.
The present case is being reported for the rarity of the syndrome.
| 1. |
Badame AJ. Review of progeria. Arch Dermatol 1989;125:540-4.
[Google Scholar]
|
| 2. |
DeBusk FL. The Hutchinson-Gilford progeria syndrome. Report of 4 cases and review of the literature. J Pediatr 1972;80:697-724.
[Google Scholar]
|
| 3. |
Eriksson M, Brown WT, Gordon LB, Glynn MW, Singer J, Scott L, et al. Recurrent de novo point mutation in lamin A cause Hutchinson-Gilford progeria syndrome. Nature 2003;423:293-8.
[Google Scholar]
|
| 4. |
Burrows NP, Lovell CR. Disorders of connective tissue. In: Burns T, Breathnach S, Cox N, Griffiths C, editors. Rook's Textbook of Dermatology. 7 th ed. Vol. 4. London: Blackwell Science Ltd; 2004. p. 49-50.
th ed. Vol. 4. London: Blackwell Science Ltd; 2004. p. 49-50.'>[Google Scholar]
|
| 5. |
Hamer L, Kaplan F, Fallon M. The musculoskeletal manifestations of progeria: A literature review. Orthopedics 1988;11:763-9.
[Google Scholar]
|
Fulltext Views
5,734
PDF downloads
1,877





![[Figure - 1]](#fig_ijdvl_2010_76_5_591_69094_f1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2010_76_5_591_69094_f2.jpg){kind=link}
![[Figure - 3]](#fig_ijdvl_2010_76_5_591_69094_f3.jpg){kind=link}
![[Figure - 4]](#fig_ijdvl_2010_76_5_591_69094_f4.jpg){kind=link}
![[Figure - 5]](#fig_ijdvl_2010_76_5_591_69094_f5.jpg){kind=link}