
Translate this page into:
Innovative drug delivery systems for leprosy treatment
Corresponding author: Prof. Nadia Araci Bou-Chacra, Lineu Prestes Avenue, 580, São Paulo, SP, Brazil, chacra@usp.br
-
Received: ,
Accepted: ,
How to cite this article: Rocha NPD, Barbosa EJ, de Araujo GLB, Bou-Chacra NA. Innovative drug delivery systems for leprosy treatment. Indian J Dermatol Venereol Leprol 2022;88:437-42.
Introduction
Leprosy is one of the oldest human epidemic diseases and is still endemic in some areas.1,2 The last WHO report, from 2018, counted 208,641 new cases globally, with India, Brazil and Indonesia concentrating approximately 80% (166,011) of them.2 Dapsone, rifampicin and clofazimine constitute the multidrug therapy, which has reduced new leprosy cases remarkably, since its inception in the 1980s.3-5 According to the clinical type of disease, treatment duration varies from 6 to 12 months.6
Low adherence to therapy is one of the main hurdles for leprosy elimination as the disease requires prolonged treatment.7 A complex interplay of factors such as socioeconomic condition, inadequate healthcare service, and multidrug therapy underlie the poor adherence.7 Among the factors associated with multidrug therapy drugs, resistance and adverse effects are important.8-10 The first extensive study from endemic countries revealed that 8.0% of M. leprae strains underwent mutations, resulting in multidrug therapy resistance.9 However, a comprehensive study addressing drug adverse effects is still absent. Notably, a retrospective study from Brazil reported at least one adverse effect related to multidrug therapy in almost 37.9% patients.11
An alternative treatment to WHO-multidrug therapy consists of rifampicin, ofloxacin and minocycline, called ROM. This alternate regimen utilizes the bactericidal/bacteriostatic activity of both drugs, ofloxacin and minocycline.12 Although the resistance rate to ofloxacin (1.3%) is relevant, ROM-drugs usually present mild adverse effects.9,13
Most drugs used for leprosy treatment have low water solubility, which limits their bioavailability.14,15,16 Accordingly, administration of high doses required for reaching therapeutic blood levels aggravate adverse effects. Poor water solubility of these drugs may also result in their variable serum concentration, thus increasing the likelihood of bacterial resistance.17-19 Additionally, rifampicin and clofazimine bioavailability may be limited, respectively, by stomach degradation and recrystallization depending on pH.11,20,21 Unlike other drugs, minocycline is highly water soluble, its major limitation being intestinal permeability.22
New formulations have been proposed for leprosy therapy to address these problems. This article aims to highlight the recent advances in drug delivery systems, which may be utilized to overcome these hurdles.
Innovative pharmaceutical strategies towards enhancement of therapeuticefficacy
Recent advances in drug delivery systems may overcome solubility impairment, common in pharmaceutical development.23 The oral formulations proposed for multidrug therapy drugs are focused on two main strategies: increasing the apparent drug water solubility or modifying the drug release.
Figure 1 summarizes the improvements and the expected results of multidrug therapy innovative formulations. In vitro evaluation, in vivo, and in silico performances of these preparations are provided in Table 1, if available, showing their potential therapeutic efficacy. As the following steps, well-established clinical tests will play a key role in ensuring their relevance for patients. Aiming to reach the market, a collaborative effort between government and private companies is essential.
| DS | Innovative formulations | In vitro evaluation (Drug release/Solubility assessment) | In vivo/in silico/in vitro performance | Ref. |
|---|---|---|---|---|
| DAP | Solid dispersion (SD) | Drug release was nearly 1.9-fold compared to physical mixture and 7.5-fold compared to pure DAP (in first 10 min). | - | 24 |
| DAP and CFZ | Polymeric nanoparticles | Sustained release: after 24 h, 82% of DAP and 68% of CFZ. | NP was more effective than the same dose of the drugs. | 43 |
| DAP | Cocrystal | Best solubility achieved: 1.5 times, compared to pure DAP. | - | 26 |
| DAP | Hydrogel | In the first hours, up to 5%, after 4 h 10% and sustained release (up to 20%) in the next 22 h. | - | 44 |
| DAP | Nanofibers | After 400 min, 77.71%, compared to 80.61% of DAP nanoemulsion. | - | 45 |
| DAP | Salt and Eutectics | Dissolution rate of salt nearly 2-fold and eutectics 1.7-fold than pure DAP (in first 10 min). | - | 25 |
| RIF | Solid lipid nanoparticles (SLN) | Drug release was 70.12% after 9 days while free RIF was more than 90% in 24 h. |
in plasma, SLN: 15.12 µg/ml, Free RIF: 2.27 µg/ml. Relative bioavailability was improved 8.16 times (compared to free RIF), with sustained levels for 5 days.max studies: CIn vivo |
17 |
| RIF | Particulate hydrogel beads | Constant and sustained drug level throughout 24 h, with highest amount of drug released of 71.49%. | - | 49 |
| RIF | Solid dispersion | Drug release was 82.3%, compared to 32.7% of RIF powder (at 60 min, pH 6.8). | - | 28 |
| RIF | Solid lipid nanoparticle (SLN) | 85% within approximately 6 min at both pHs performed (1.2 and 6.8). |
increased by 3.72 and 5.22-fold compared to the RIF suspension. GastroPlus™ predicted maximum compartmental absorption from proximal and distal portions of the intestine.max assessment: AUC and CIn vivo-in silico |
18 |
| RIF | Chitosan/gelatin/lecithin nanoparticles | Drug release was more than 3-fold up compared to free drug, at higher concentration of lecithin (2.0 g), in pH 7.2. | - | 48 |
| RIF | Carboxymethylcellulose complex | Drug release was 99 ± 3% at 15 min, compared to two commercial medicines of less than 80% and approximately 90%. | - | 33 |
| RIF | Phospholipid lipospheres | The best formulation presented solubility of 350.9 ± 23 μg/mL compared to 105.1 ± 12 μg/mL of pure drug. | Antimycobacterial activity enhanced compared to pure drug. ) was 109.92 ± 25 μg/mL compared to 54.31 ± 18 μg/mL of pure drug.maxPeak plasma concentration (C The AUC was 406.92 ± 18 μg h/L compared to 147.72 ± 15 μg h/L of pure drug. |
29 |
| RIF | DIMEB complex | Improved solubility, at pH 7.4, achieving the equilibrium in approximately 9 h. | - | 20 |
| RIF | Solid lipid nanoparticles (SLN) | Biphasic profile: initial burst followed by sustained pattern (up to 90% drug in 120 h). | - | 46 |
| RIF | Niosome | Between 61.69% and 75.90%, compared to 32.43% of pure RIF (after the first 2 h). | - | 30 |
| RIF | Co-polymeric nanoparticles (NP) | Solubility improved 65-fold compared to pure drug. Controlled release achieving up to 70 h, compared to 6 h of pure drug. | - | 47 |
| RIF | Niosome | Achieving 80% of drug release compared to 40% of pure drug, over 12 h. | - | 31 |
| RIF | Liposomes | Drug release achieved 95% released after only 5 h, compared to nearly 70% of free drug. | - | 32 |
| RIF | Nanocrystals | Nanocrystals showed up to 1.74-fold on solubility compared to commercial product. | - | 27 |
| CFZ | Alginate-mediated carrier | The release rate decreases upon increasing alginate concentrations. | - | 58 |
| CFZ | Complex formation | Increased approximately 0.53-fold of the maximum solubility compared to CB[7]. | The analysis of MIC50 between complex and free drug did not show significant statistical difference. | 38 |
| CFZ | Nanoporous silica particles | Solubility was increased by 20-fold in simulated gastric fluid. | Permeation studies (using Caco-2 intestinal cells) showed more than 5-fold increased intestinal permeation in comparison to the free drug (below the detection limit). | 37 |
| CFZ | Enzyme-mediated carrier | Only CFZ binded to pepsin remains in solution in the intestinal environment (pH~5.4) | - | 56 |
| CFZ | Polymeric nanoparticle | Sustained pattern, about 30% at the end of the experiment (buffer solution - pH 6.8 for 8 h, at 37°C). | - | 42 |
| CFZ | Mesoporous silica particles (MPS) | At pH 4.1, maximum of 29% for ho-MSP (more hydrophobic) and 46% for hi-MSP. At pH 6.8: rapid release from hi-MSP, with 2 times higher initial release, compared to ho-MSP. However, both released nearly 10%. | - | 57 |
| CFZ | Nanoparticle (NP) |
.®The NP, mainly in presence of fat, was faster dissolved compared to drug substance or to Lamprene |
36 | |
| CFZ | Salt | Improvement of 5-fold on solubility compared to the free drug. | - | 35 |
| OFL | Cocrystal salt | After 1h, the amount of dissolved from cocrystal was more than 3-fold, compared to pure drug. | - | 39 |
| OFL | Inclusion complex | Solubility increased 3.7-fold compared to pure drug. | - | 40 |
| OFL | Nanoparticle cellulose conjugates | Nanoparticles showed sustained release proved in a pharmacokinetic study. | AUC was 1.6-2.3 times higher than controls rabbits. | 59 |
| OFL | Nanofibres | Initial rapid release (>50% of drug released within 4 h), followed by a slow and sustained release phase. |
mucoadhesion and gastro-retention in rats.in vivo antimicrobial activity, and in vitro Enhanced |
60 |
| OFL | Polymeric complex | Release of 45-57% in 50 h. |
microbiological studies.in vitro in M. tuberculosis Formulations demonstrated activity against |
61 |
| OFL | Nanoparticle (NP) | Maximum drug release of 76% observed after 18 h (ph 2.2). |
.E. coli Proven antibacterial activity against |
62 |
| OFL | PEGylated nanoparticle (NP) | The best nanoparticles obtained released 96% of OFL in 36 h. Free drug was released 100% in less than 4 h. |
resistance.Bacillus subtilis Better bactericidal activity compared to free drug. And inhibition of |
63 |
| MINO | Hydrogel | Initial burst release with subsequent release control, achieving 100% only after more than 48 h. | - | 64 |
| MINO | Nanoparticle | Drug release achieved nearly 90% only after more than 10h. Free drug achieve the same percentage within 1 h. | Antimicrobial activity was comparable to the free drug. | 65 |
| MINO | Solid lipid nanoparticle (SLN) |
release was kept continuous during 7 days.In vitro |
SLN was twice as efficient as free drug, in animal tests. | 66 |
| MINO | Polymeric nanoparticles | The NP presented an initial burst during 24h and a linear release over 30 days, compared to more than 98% within two days of free drug. | - | 67 |
DS: Drug substance; Ref: References; DAP: Dapsone; RIF: Rifampicin; CFZ: Clofazimine; OFL: Ofloxacin; MINO: Minocycline; h: hours; AUC: Area under the curve; DIMEB: Heptakis(2,6-di-O-methyl)-β-cyclodextrin
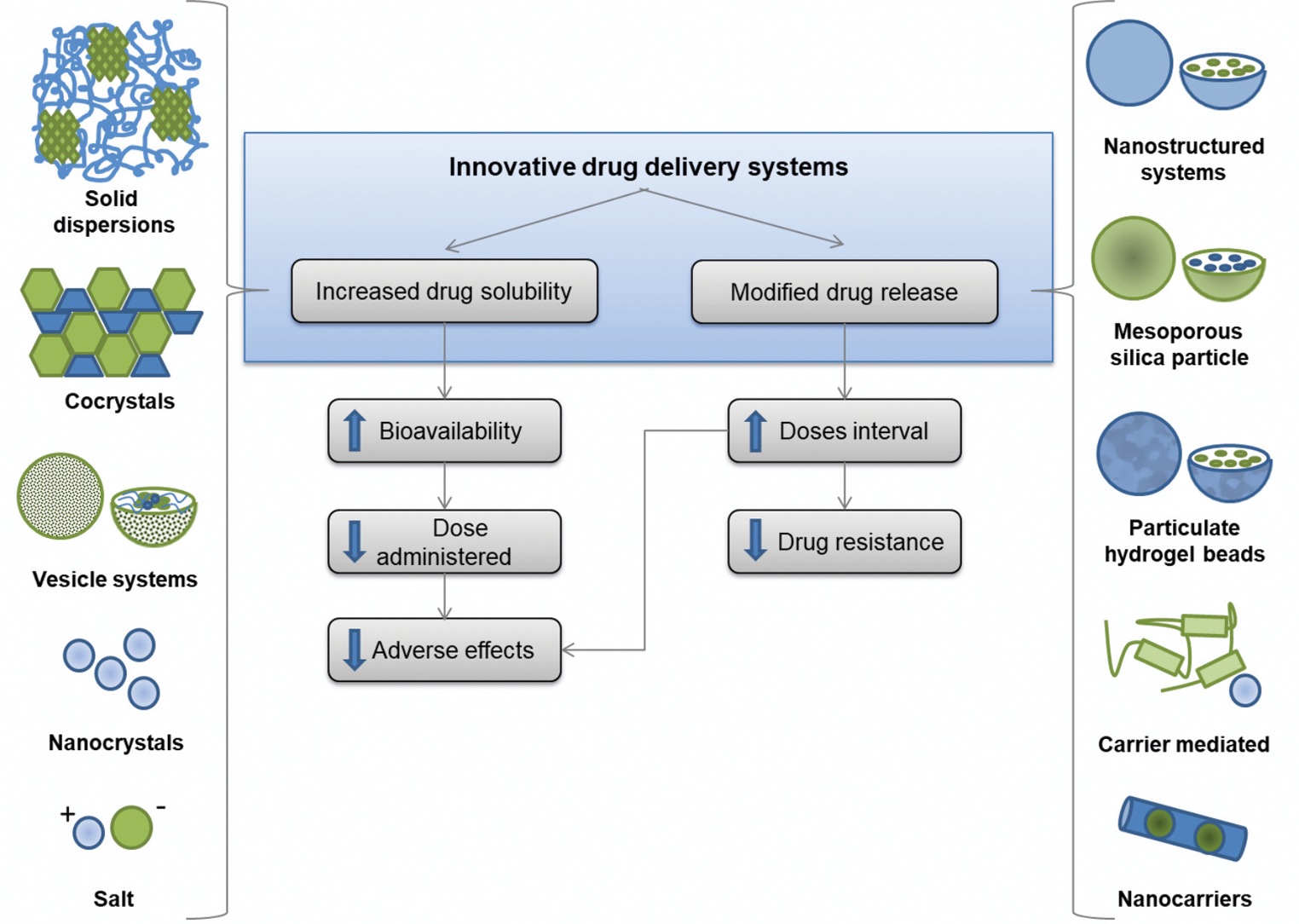
- Innovative drug delivery systems formulations developed to overcome the main hurdles in the treatment of leprosy
Drug water solubility enhancement
For dapsone, innovative formulations obtained through synthesis of its chemical derivatives (salt and cocrystal) and solid dispersion were proposed to enhance water solubility.24-26 For example, solid dispersion increased water solubility by more than 7.5-fold, compared to free drug form.24 The improved bioavailability may reduce the therapeutic dose.
The solubility of rifampicin has been improved by using nanocrystals, solid dispersion, vesicle systems (liposphere, niosome and liposome), and complex preparations.20,27-33 Rifampicin nanocrystals enable 2-fold increase of drug concentration in a formulation. This innovative preparation halves the original intended dose.27 Apart from dose reduction, nanocrystals may increase permeation into intestine cells due to enhanced adherence.27 Rifampicin absorption may be reduced to half by food, which may also be mitigated by nanocrystals.27,34
Clofazimine innovative preparations include synthetic chemical derivatives (salt and complex) and nanotechnology-based delivery (nanoparticle and nanoporous silica particle).35-38 Amongst them, nanoporous silica particles increased its water solubility and intestinal permeability by 20-fold and 5-fold, respectively, compared to free-form.37 These modifications can significantly reduce the effective therapeutic dose.
The formulations proposed to enhance ofloxacin solubility are cocrystal and cyclodextrin complex.39,40 Initially, such formulations were developed for ophthalmic topical preparations. Minocycline does not present aqueous solubility issues,22 instead, newer strategies such as release modifications are aiming to improve its permeability.
Modified drug release
Modified release strategy aims to modulate drug release from the dosage form. For instance, enteric release is designed for intestinal drug delivery, protecting it from gastric pH. The extended-release formulations restrict drug delivery immediately following oral administration.29,30 The enteric release is especially relevant for rifampicin and clofazimine due to their chemical instability in acidic conditions.41,42
Dapsone modified-release was proposed using different strategies such as polymeric nanoparticles, hydrogels and nanofibers.43-45 In vivo results of polymeric nanoparticles demonstrated the sustained co-delivery of dapsone and clofazimine could reduce their doses.43 For rifampicin, strategies included nanoparticles (solid lipid, polymeric and lecithin), complex, and hydrogel beads.17,18,46,47 A formulation presenting an initial burst followed by sustained release is desirable, as observed in over 65% (4 of 6) (Table 1) of proposed rifampicin studies.46-49 Also, studies have shown favourable in vivo or in silico results. Among these, the increase in peak plasma concentration (Cmax) is up to seven times higher than the free drug,17,18 using solid lipid nanoparticle strategy.17 Furthermore, rifampicin plasma concentration was sustained above the minimum inhibitory concentration (MIC) for five days, compared to two days of free drug (Table 1). Thus, these formulations can reduce the dose and minimize adverse effects.17
Nano-based drug delivery systems have been approved by regulatory agencies and prescribed in the last decades, reinforcing their efficacy and safety.50,51 For instance, liposomal amphotericin B (AmBisome) has been used to treat leishmaniasis successfully.52 Nevertheless, the particle size and shape can impact the nanoparticle distribution.53 For example, nanorods may accumulate in organs related to immune response and blood clearance, such as lymph nodes, spleen, liver and bone marrow.54 Consequently, risk assessment and quantification methods have been increasingly explored aiming to evaluate nanomedicine effects for patients.54,55
Clofazimine, in turn, can recrystallize outside a pH range of 2-4, thereby compromising absorption.56 Polymeric nanoparticles, alginate, pepsin and mesoporous silica are examples of carriers developed to modify the release of clofazimine.37,42,56-58 Polymeric nanoparticles ensured sustained release and lower cellular toxicity in Caco-2 and HT29-MTX cells, compared to free drug. Thus, these studies corroborated the success of this strategy to avoid clofazimine recrystallization.42
Strategies for ofloxacin included cellulose conjugate, nanofibers, polymeric complex and nanoparticles.59-63 In vivo studies using nanofibers depicted its role as ofloxacin reservoir, increasing its residence time in the gastrointestinal tract. Besides, an in vitro study showed significant mucoadhesion using a strip of rat’s gastric mucosal membrane and improved efficacy against micro-organisms, such as E. coli, E. faecalis, S. aureu, and P. aeruginosa. Therefore, these studies represent advanced formulations with a better oral absorption profile in leprosy treatment.60
Formulations containing minocycline were mainly developed for topical application, focused on periodontal diseases. However, the modified release approach might be considered to overcome its permeability issue. Hydrogel, nanoparticles, solid lipid nanoparticles and polymeric nanoparticles have been proposed to achieve this goal.64-67
Conclusion
Leprosy elimination involves a series of treatment-related challenges, leading to poor patient adherence. The multidrug therapy drugs are distributed free of charge; however, their low water solubility, severe adverse effects and resistance potential limit treatment completion. Innovative drug delivery systems are being proposed to overcome these limitations, involving two main targets: water solubility improvement and sustained drug release. Enhanced solubility may reduce the administered dose to patient, thus minimizing adverse effects. A modified drug release approach may increase dose interval, reducing the occurrence of bacterial resistance and adverse effects. These modifications may improve patient adherence to treatment, diminishing bacillary spread by untreated patients. A reinvention of leprosy treatment may promote patient healing and interrupt transmission, two essential goals towards a leprosy-free world.
Declaration of patient consent
Patient’s consent is not required as there are no patients in this study.
Financial support and sponsorship
This work was supported by CAPES (Coordenação de Aperfeiçoamento de Pessoal de Nível Superior) - Finance code 001.
Conflicts of interest
There are no conflicts of interest.
References
- Leprosy - An overview of clinical features, diagnosis, and treatment. JDDG - J Ger Soc Dermatology. 2017;15:801-27.
- [CrossRef] [PubMed] [Google Scholar]
- Weekly Epidemiological Record - Global Leprosy Update Moving towards a Leprosy-Free World; 2018. 2019.
- [Google Scholar]
- Phylogenomics and antimicrobial resistance of the leprosy bacillus Mycobacterium leprae. Nat Commun. 2018;9:1-11.
- [CrossRef] [PubMed] [Google Scholar]
- The impact of multidrug therapy on the epidemiological pattern of leprosy in Juiz de Fora, Brazil. Cad Saúde Pública. 2000;16:343-50.
- [CrossRef] [PubMed] [Google Scholar]
- Progress and impact of multidrug therapy (MDT) implementation to leprosy control in Thailand. Japanese J Lepr. 1995;64:214-9.
- [CrossRef] [PubMed] [Google Scholar]
- Challenges beyond elimination in leprosy. Int J Mycobacteriology. 2017;6:222-8.
- [CrossRef] [PubMed] [Google Scholar]
- Interruption and defaulting of multidrug therapy against leprosy: Population-based study in Brazil’s Savannah region. PLoS Negl Trop Dis. 2011;5:4-9.
- [Google Scholar]
- Geographic and socioeconomic factors associated with leprosy treatment default: An analysis from the 100 Million Brazilian Cohort. PLoS Negl Trop Dis. 2019;13:1-18.
- [Google Scholar]
- Antimicrobial resistance in leprosy: results of the first prospective open survey conducted by a WHO surveillance network for the period 2009-15. Clin Microbiol Infect. 2018;24:1305-10.
- [CrossRef] [PubMed] [Google Scholar]
- Adverse effects from multi-drug therapy in leprosy: A Brazilian study. Lepr Rev. 2007;78:216-22.
- [PubMed] [Google Scholar]
- Efeitos adversos da poliquimioterapia em pacientes com hanseníase: um levantamento de cinco anos em um Centro de Saúde da Universidade Federal de Uberlândia. Rev Soc Bras Med Trop. 2005;35:453-60.
- [Google Scholar]
- Second-line anti-leprosy drugs: Indian experience. Indian J Drugs Dermatology. 2020;6(1):1-4.
- [CrossRef] [Google Scholar]
- 4,4’-Diaminodiphenyl sulphone: solubility and distribution in blood. Biochem J. 1962;83:417-20.
- [CrossRef] [PubMed] [Google Scholar]
- Implication of biopharmaceutics and pharmacokinetics of rifampicin in variable bioavailability from solid oral dosage form. Biopharm Drug Dispos. 2005;26:321-34.
- [CrossRef] [PubMed] [Google Scholar]
- Clofazimine mesylate: A high solubility stable salt. Cryst Growth Des. 2012;12:6250-9.
- [CrossRef] [Google Scholar]
- Nano-formulation of rifampicin with enhanced bioavailability: Development, characterization, and in-vivo safety. Int J Pharm. 2015;485:138-51.
- [CrossRef] [PubMed] [Google Scholar]
- In vitro - In vivo - In silico simulation studies of anti-tubercular drugs doped with a self nanoemulsifying drug delivery system. RSC Adv. 2016;6:93147-61.
- [CrossRef] [Google Scholar]
- Development and characterization of a new oral dapsone nanoemulsion system: permeability and in silico bioavailability studies. Int J Nanomedicine. 2012;7:5175-82.
- [CrossRef] [PubMed] [Google Scholar]
- Formation, characterization and pH dependence of rifampicin: heptakis(2,6-di-O-methyl)-β-cyclodextrin complexes. Int J Pharm. 2017;531:668-75.
- [CrossRef] [PubMed] [Google Scholar]
- Delivery of a hydrophobic drug into the lower gastrointestinal system via an endogenous enzyme-mediated carrier mechanism: An in vitro study. Eur J Pharm Biopharm. 2018;133:12-9.
- [CrossRef] [PubMed] [Google Scholar]
- Applying Biopharmaceutical Classification System (BCS) Criteria to Predict Oral Absorption of Drugs in Dogs: Challenges and Pitfalls. AAPS J. 2015;17(4):948-64.
- [CrossRef] [PubMed] [Google Scholar]
- Strategies for formulating and delivering poorly water-soluble drugs. J Drug Deliv Sci Technol. 2015;30:342-51.
- [CrossRef] [Google Scholar]
- Rational and precise development of amorphous polymeric systems with dapsone by response surface methodology. Int J Biol Macromol. 2015;81:662-71.
- [CrossRef] [PubMed] [Google Scholar]
- Eutectics and salt of dapsone with hydroxybenzoic acids: Binary phase diagrams, characterization, and evaluation. J Pharm Sci. 2020;109:2224-36.
- [CrossRef] [PubMed] [Google Scholar]
- Drug-polymer co-crystals of dapsone and polyethylene glycol: An emerging subset in pharmaceutical cocrystals. Cryst Growth Des. 2018;18:7590-8.
- [CrossRef] [Google Scholar]
- Rifampicin nanocrystals: Towards an innovative approach to treat tuberculosis. Mater Sci Eng C. 2020;112:110895.
- [CrossRef] [Google Scholar]
- Impact of HPMC on the physical properties of spray-congealed PEG microparticles and its swelling effect on rifampicin dissolution. Drug Dev Ind Pharm. 2016;42:403-11.
- [CrossRef] [PubMed] [Google Scholar]
- In vitro-in vivo evaluation of novel co-spray dried rifampicin phospholipid lipospheres for oral delivery. AAPS PharmSciTech. 2017;18:138-46.
- [CrossRef] [PubMed] [Google Scholar]
- Process optimization of ecological probe sonication technique for production of rifampicin loaded niosomes. J Drug Deliv Sci Technol. 2019;50:27-33.
- [CrossRef] [Google Scholar]
- Formulation optimization and in vitro characterization of rifampicin and ceftriaxone dual drug loaded niosomes with high energy probe sonication technique. J Drug Deliv Sci Technol. 2020;58:101763.
- [CrossRef] [Google Scholar]
- Simultaneous liposomal encapsulation of antibiotics and proteins: Co-loading and characterization of rifampicin and Human Serum Albumin in soy-liposomes. J Drug Deliv Sci Technol. 2020;58:101751.
- [CrossRef] [Google Scholar]
- Very fast dissolving acid carboxymethylcellulose-rifampicin matrix: Development and solid-state characterization. Eur J Pharm Sci. 2017;96:398-410.
- [CrossRef] [PubMed] [Google Scholar]
- Rifampin (INN Rifampicin) J Pain Symptom Manage. 2015;50:891-95.
- [CrossRef] [PubMed] [Google Scholar]
- A new salt of clofazimine to improve leprosy treatment. J Mol Struct 2020:1214.
- [CrossRef] [Google Scholar]
- Solid-state behavior and solubilization of flash nanoprecipitated clofazimine particles during the dispersion and digestion of milk-based formulations. Mol Pharm. 2019;16:2755-65.
- [CrossRef] [PubMed] [Google Scholar]
- Clofazimine encapsulation in nanoporous silica particles for the oral treatment of antibiotic-resistant Mycobacterium tuberculosis infections. Nanomedicine. 2017;12:831-44.
- [CrossRef] [PubMed] [Google Scholar]
- Complexation of clofazimine by macrocyclic cucurbit[7]uril reduced its cardiotoxicity without affecting the antimycobacterial efficacy. Org Biomol Chem. 2016;14:7563-9.
- [CrossRef] [PubMed] [Google Scholar]
- Improving solubility and intrinsic dissolution rate of ofloxacin API through salt formation via mechanochemical synthesis with diphenic acid. J Mol Struct. 2020;1221:128806.
- [CrossRef] [Google Scholar]
- Study on a host-guest interaction of hydroxypropyl-β-cyclodextrin with ofloxacin. J Mol Liq. 2015;202:101-6.
- [CrossRef] [Google Scholar]
- Rifampin stability and solution concentration enhancement through amorphous solid dispersion in cellulose ω-carboxyalkanoate matrices. J Pharm Sci. 2018;107:127-38.
- [CrossRef] [PubMed] [Google Scholar]
- Development of PLGA nanoparticles loaded with clofazimine for oral delivery: Assessment of formulation variables and intestinal permeability. Eur J Pharm Sci. 2018;112:28-37.
- [CrossRef] [PubMed] [Google Scholar]
- Nano carrier mediated co-delivery of dapsone and clofazimine for improved therapeutic efficacy against tuberculosis in rats. Biomed Res. 2017;28:1284-9.
- [Google Scholar]
- pH-responsive chitosan based hydrogels affect the release of dapsone: Design, set-up, and physicochemical characterization. Int J Biol Macromol. 2019;133:1268-79.
- [CrossRef] [Google Scholar]
- Preparation of polyacrylamide/polylactic acid co-assembled core/shell nanofibers as designed beads for dapsone in vitro efficient delivery. Artif Cells, Nanomedicine Biotechnol. 2019;47:917-26.
- [CrossRef] [PubMed] [Google Scholar]
- Formulation, optimization, and characterization of rifampicin-loaded solid lipid nanoparticles for the treatment of tuberculosis. Drug Dev Ind Pharm. 2018;44:1975-89.
- [Google Scholar]
- HPMA-PLGA based nanoparticles for effective in vitro delivery of rifampicin. Pharm Res. 2019;36:1-12.
- [CrossRef] [Google Scholar]
- Modified rifampin nanoparticles: Increased solubility with slow release rate. Int J Mycobacteriology. 2017;6:171.
- [CrossRef] [PubMed] [Google Scholar]
- Natural Gellan gum (Phytagel®) based novel hydrogel beads of rifampicin for oral delivery with improved functionality. Indian J Pharm Educ Res. 2016;50:S159-67.
- [CrossRef] [Google Scholar]
- Nanomedicine(s) under the microscope. 2011 Dec 5. Mol Pharm. 8:2101-41. Available from: https://pubs.acs.org/doi/10.1021/mp200394t.
- [CrossRef] [Google Scholar]
- The regulation of nanomaterials and nanomedicines for clinical application: Current and future perspectives. Biomater Sci. 2020;8(17):4653-64.
- [CrossRef] [Google Scholar]
- Liposomal amphotericin B as a treatment for human leishmaniasis. Expert Opin Emerg Drugs. 2012;17:493-510.
- [CrossRef] [Google Scholar]
- The impact of size and surface ligand of gold nanorods on liver cancer accumulation and photothermal therapy in the second near-infrared window. 2020. J Colloid Interface Sci. 565:186-96. Available from: https://doi.org/10.1016/j.jcis.2020.01.026.
- [CrossRef] [PubMed] [Google Scholar]
- Quantification of gold nanoparticle accumulation in tissue by two-photon luminescence microscopy. Nanoscale. 2019;11:11331-9.
- [CrossRef] [Google Scholar]
- Risk assessment and risk minimization in nanomedicine: A need for predictive, alternative, and 3Rs strategies. Front Pharmacol. 2018;9:1-7.
- [CrossRef] [PubMed] [Google Scholar]
- Delivery of a hydrophobic drug into the lower gastrointestinal system via an endogenous enzyme-mediated carrier mechanism: An in vitro study. Eur J Pharm Biopharm. 2018;133:12-19.
- [CrossRef] [PubMed] [Google Scholar]
- Fluorescence imaging of antibiotic clofazimine encapsulated within mesoporous silica particle carriers: Relevance to drug delivery and the effect on its release kinetics. Phys Chem Chem Phys. 2018;20:11899-911.
- [CrossRef] [Google Scholar]
- Amphiphilic alginate as a drug release vehicle for water-insoluble drugs. Colloid J. 2015;77:754-60.
- [CrossRef] [Google Scholar]
- Cellulose ether derivatives: A new platform for prodrug formation of fluoroquinolone antibiotics. Cellulose. 2015;22:2011-22.
- [CrossRef] [Google Scholar]
- Ofloxacin loaded gellan/PVA nanofibers - Synthesis, characterization, and evaluation of their gastroretentive/mucoadhesive drug delivery potential. Mater Sci Eng C. 2017;71:611-9.
- [CrossRef] [PubMed] [Google Scholar]
- Polymeric complexes of ofloxacin and their activity against tuberculosis mycobacteria. Pharm Chem J. 2017;51(4):250-3.
- [CrossRef] [Google Scholar]
- Fabrication of chitosan-g-poly(acrylamide)/CuS nanocomposite for controlled drug delivery and antibacterial activity. Mater Sci Eng C. 2016;64:428-35.
- [CrossRef] [PubMed] [Google Scholar]
- PEGylated ofloxacin nanoparticles render strong antibacterial activity against many clinically important human pathogens. Colloids Surfaces B Biointerfaces. 2015;132:62-70.
- [CrossRef] [PubMed] [Google Scholar]
- MMP-8-Responsive polyethylene glycol hydrogel for intraoral drug delivery. J Dent Res. 2019;98:564-71.
- [CrossRef] [PubMed] [Google Scholar]
- Minocycline and silver dual-loaded polyphosphoester-based nanoparticles for treatment of resistant pseudomonas aeruginosa. Mol Pharm. 2019;16:1606-19.
- [CrossRef] [PubMed] [Google Scholar]
- Antinociceptive antibiotics-loaded into solid lipid nanoparticles of prolonged release: Measuring pharmacological efficiency and time span on chronic monoarthritis rats. PLoS One. 2018;13:1-15.
- [CrossRef] [PubMed] [Google Scholar]
- Hydrophobic ion pairing of a minocycline/Ca2+/AOT complex for preparation of drug-loaded PLGA nanoparticles with improved sustained release. Int J Pharm. 2016;499:351-7.
- [CrossRef] [PubMed] [Google Scholar]




