Translate this page into:
Laugier-Hunziker syndrome
Correspondence Address:
Dinesh P Asati
Department of Dermatology, L. N. Medical College and JK Hospital, Kolar Road, Bhopal - 462 042, Madhya Pradesh
India
| How to cite this article: Asati DP, Tiwari S. Laugier-Hunziker syndrome. Indian J Dermatol Venereol Leprol 2011;77:536 |
Sir,
Idiopathic lenticular mucocutaneous pigmentation [Laugier-Hunziker syndrome (LHS)] is a benign pigmentary disorder characterized by discrete and confluent, slate to brown-black macules on skin and mucosae. [1],[2] It is a rare disorder which represents an important clinical differential to Peutz-Jeghers syndrome (PJS), which is associated with gastrointestinal hamartomatous polyps and various benign and malignant neoplasms. Absence of these systemic findings suggests the diagnosis of LHS. [3]
A 19-year-old female presented with multiple, discrete, brown-black oval to irregular shaped macules on lips, buccal mucosae, palate, tongue, and perioral skin, since birth. She also developed multiple light brown freckles on upper part of face including periorbital skin, nose, cheeks, fingertips and nail folds at the age of 10-12 years, which increased insidiously in number for the next 4-5 years during adolescence and since then it remained static. She had normal built and no history suggestive of intestinal involvement (like recurrent abdominal pain, vomiting, gastrointestinal bleed, malena), palpitation or weakness. There was no history of any drug intake or excessive sun exposure. Rest of the skin was normal and there were no blue nevi, soft tissue swellings or hair abnormalities. There was no family history of similar skin lesions or findings suggestive of gastrointestinal involvement seen in PJS.
Examination revealed multiple brownish black lenticular macules on perioral skin, lips, buccal mucosae, tongue and palate. Many tiny light brown irregular and angulated macules were suggestive of freckles and they were prominently present on nose, periorbital skin, cheeks and lower forehead [Figure - 1]. Similar irregular shaped freckles were also found on the fingertips and nail folds. Longitudinal melanonychia was present in the form of streaks of hyperpigmented macules on the nail plates as well. Ocular mucosa, genitalia and toenails were spared. Rest of skin, mucosae and hair examination was normal. Routine hematological and biochemical parameters were within normal reference range. Intestinal polyposis was ruled out by barium meal follow-through study. Invasive (endoscopic) evaluation of gastrointestinal tract was withheld in view of absence of any symptoms suggestive of intestinal involvement; also, the patient did not give consent for endoscopy. Biopsy from one lentigen on lip showed basal hyperpigmentation and pigment incontinence. An exclusion diagnosis of LHS was made; the patient and her family members were reassured about the benign nature of the disease.
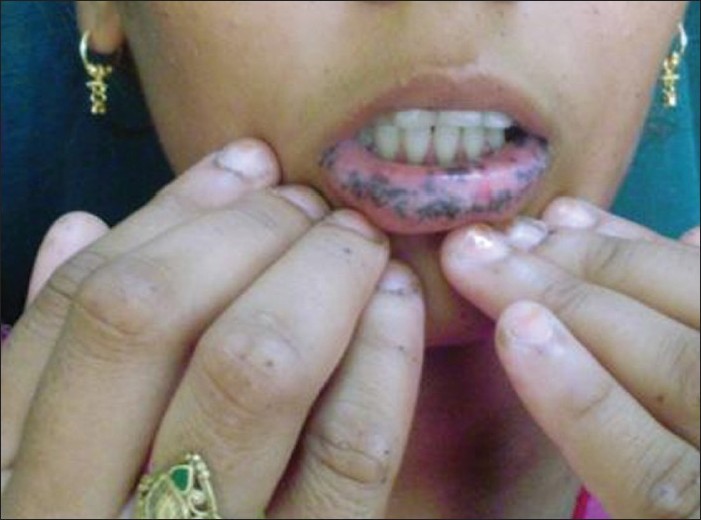 |
| Figure 1: Involvement of lips, fingers and longitudinal melanonychia in the patient |
LHS is characterized by asymptomatic, discrete to confluent, hyperpigmented lenticular mucocutaneous macules of slate to brown-black color. Lips and oral cavity including buccal mucosae, tongue and gingivae are most commonly involved, but involvement of fingers, nails, palms, toes and external genitalia is also described. [2],[3] There is increased epidermal melanin content with or without increase in melanocytes and presence of macrophages and pigment incontinence in the papillary dermis on histopathology. [4],[5] Rete ridges may be normal or elongated in size. Basement membrane is intact and nevus cells are not seen. Important clinical differentials of mucocutaneous hyperpigmentation include PJS, LEOPARD syndrome, LAMB syndrome, McCune-Albright syndrome, Addison disease and Gardner syndrome. [3] These conditions were ruled out by history and clinical examination. We did not subject the patient to extensive invasive investigations or genetic studies, which were also not justified in the absence of clinical clue toward PJS or other pigmentary disorders. One unusual feature in our case was the congenital onset of pigmentation as LHS usually has adult onset. Since we did not perform molecular genetic studies, complete exclusion of PJS could not be done.
Q-switched alexandrite laser has been reported to be effective and safe for treating pigmented lesions of LHS. [6] Physicians should be aware of LHS as an important clinical differential of other serious pigmentary disorders like PJS to avoid unnecessary use of intensive malignancy screening protocols.
| 1. |
Moore RT, Chae KA, Rhodes AR. Laugier and Hunziker pigmentation: A lentiginous proliferation of melanocytes. J Am Acad Dermatol 2004;50:70-4.
[Google Scholar]
|
| 2. |
Sardana K, Mishra D, Garg V. Laugier-Hunziker syndrome. Indian Pediatr 2006;43:998-1000.
[Google Scholar]
|
| 3. |
Lampe AK, Hampton PJ, Woodford-Richens K, Tomlinson I, Lawrence CM, Douglas FS. Laugier-Hunziker syndrome: An important differential diagnosis for Peutz-Jeghers syndrome. J Med Genet 2003;40:e77.
[Google Scholar]
|
| 4. |
Gencoglan G, Turk BG, Karaarslan IK, Akalin T, Ozdemir F. Dermoscopic Findings in Laugier-Hunziker syndrome. Arch Dermatol 2007;143:631-3.
[Google Scholar]
|
| 5. |
Yago K, Tanaka Y, Asanami S. Laugier-Hunziker-Baran syndrome. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2008;106:e20-5.
[Google Scholar]
|
| 6. |
Zuo YG, Ma DL, Jin HZ, Liu YH, Wang HW, Sun QN. Treatment of Laugier-Hunziker syndrome with the Q-switched alexandrite laser in 22 Chinese patients. Arch Dermatol Res 2010;302:125-30.
[Google Scholar]
|
Fulltext Views
3,849
PDF downloads
2,086





![[Figure - 1]](#fig_ijdvl_2011_77_4_536_82422_u1.jpg){kind=link}