Translate this page into:
Lipoid proteinosis in the eastern Mediterranean region of Turkey
2 Department of Internal Medicine, School of Medicine, University of Mustafa Kemal, Hatay, Turkey
3 Department of Radiology, School of Medicine, University of Mustafa Kemal, Hatay, Turkey
Correspondence Address:
Asena C Dogramaci
Department of Dermatology, Mustafa Kemal University School of Medicine, 31100, Antakya, Hatay
Turkey
| How to cite this article: Dogramaci AC, Celik MM, Celik E, Bayarogullari H. Lipoid proteinosis in the eastern Mediterranean region of Turkey. Indian J Dermatol Venereol Leprol 2012;78:318-322 |
Abstract
Background: Lipoid proteinosis (LP), also known as hyalinosis cutis et mucosae or Urbach-Wiethe disease, is a rare autosomal recessive genodermatosis. Hyalin-like material is deposited in multiple organs, including the skin, oral mucosa, larynx, and brain. Only about 300 cases have been reported in the literature so far. Occurrence of LP in siblings is also rare. The reasons for relatively large number of cases, the clinical features of the patients, and the association of LP with other clinical conditions are described in this article. Aims: The aim of this study is to show that LP is not rare in Turkey and consanguineous marriage is still a social problem. Methods: We included patients between the years of 2008-2011 with lesions resembling LP. Based on the findings of clinical and histological examination of all cases, a diagnosis of LP was made. Results: We presented six different families with multiple family members suffered from LP. In total, we determined eight children and six adult patients. Three of eight children were from the same family (sisters), the other three children belonged to another family (brothers), two children were from another family (brother and sister), and the adult patients were from three different families. Conclusions: Patients with LP are likely to present first to a dermatologist because of the appearance of their skin; therefore, it is important that the dermatologic diagnosis is not to be missed. We described patients with LP and discuss the salient features of this disease.Introduction
Lipoid proteinosis (LP), which is also termed as hyalinosis cutis et mucosae or Urbach-Wiethe disease, is a rare autosomal recessive genodermatosis. LP was first reported by Urbach and Wiethe in 1929. [1] The true incidence and the exact pathogenesis of this disease are unknown. Homogenous hyalin-like material is deposited in multiple organs, including the skin, oral mucosa, larynx, and brain. Hoarseness is the first clinical symptom of LP in most cases. The later developments are infiltration and thickening of the skin and the mucosae. Beaded eyelid papules are the classic, easily noticeable sign. Generalized visceral involvement has also been reported. [2],[3] LP has no effective treatment so far.
There are only about 300 LP cases that have been reported in the literature. Occurrence of LP in siblings is also rare . [3],[4],[5] Herein, we reported 14 patients in six different families that include siblings, with dermatologic features consistent with LP in the Eastern Mediterranean Region of Turkey.
Methods
We included patients between the years of 2008-2011 with lesions resembling LP. All of the patients presented to the dermatology outpatient clinic for the first time. A full systemic examination was performed on patients. The abdominopelvic ultrasonography was performed on all patients for the visceral involvement. Indirect laryngoscopy was performed to all patients. Based on the findings of clinical and histological examination of all cases, a diagnosis of LP was made.
Family 1 (Patients 1-3)
A 12-year-old girl was referred to our department for itching of acneiform scars on the face and extremities. On physical examination, multiple small depressed scars varying in size from 2 to 4 mm were found on the face, arms, and legs. There were also skin-colored wart-like papules on the elbow and knees. In addition, there were beaded eyelid papules and hoarseness of voice. Family history revealed that her two sisters had similar symptoms. We also examined two of her sisters. Her 6-year-old sister had progressive laryngeal hoarseness since early infancy and multiple confluent papules on the eyelids and a lot of mild hyperpigmented, depressed scars on the face [Figure - 1]. Her 18-year-old sister had a similar problem as her younger sister. Histopathologic examination of the biopsy specimen taken from one of patient′s eyelid papules showed Periodic Acid-Schiff (PAS)-positive diffuse deposits of hyaline material.
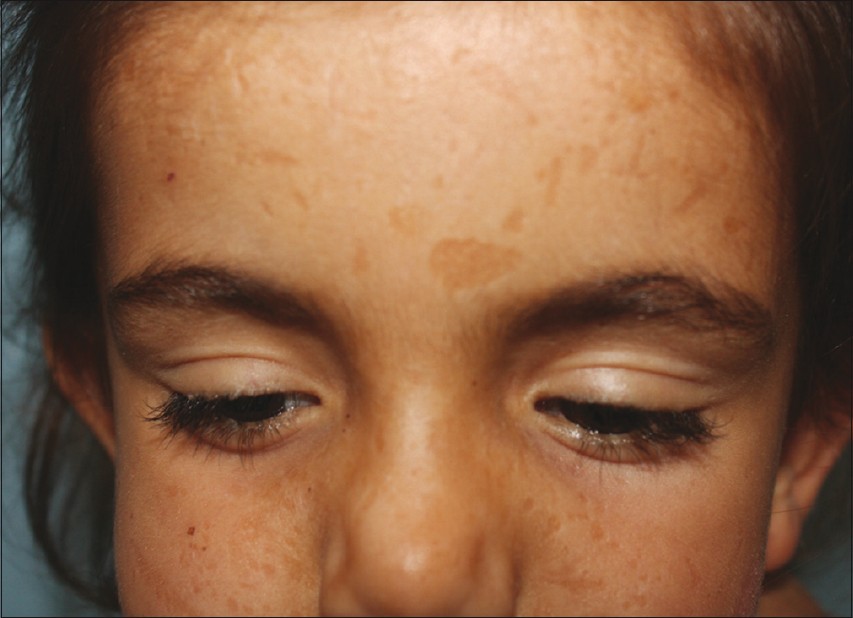 |
| Figure 1: Acneiform scars and eyelid papules on the family 1-patient 2 |
Family 2 (Patients 4-6)
A 9-year-old boy presented to our outpatient clinic with multiple depressed scars on the face and multiple bead-like papules on the eyelid margins, which had developed 7 years earlier. He had severe hoarseness, which occurred soon after birth. The skin of the elbows and armpits were thickened. Family history revealed that his two brothers (a 12-year-old and a 16-year-old) had similar skin lesions, but to a lesser degree. Microscopy of verrucous elbow lesions revealed a hyperkeratosis and mild acanthosis of the epidermis. The dermis was relatively less cellular. The appendages and capillaries showed thickening PAS-positive homogenous hyalinized material histology of these lesions that showed LP [Figure - 2].
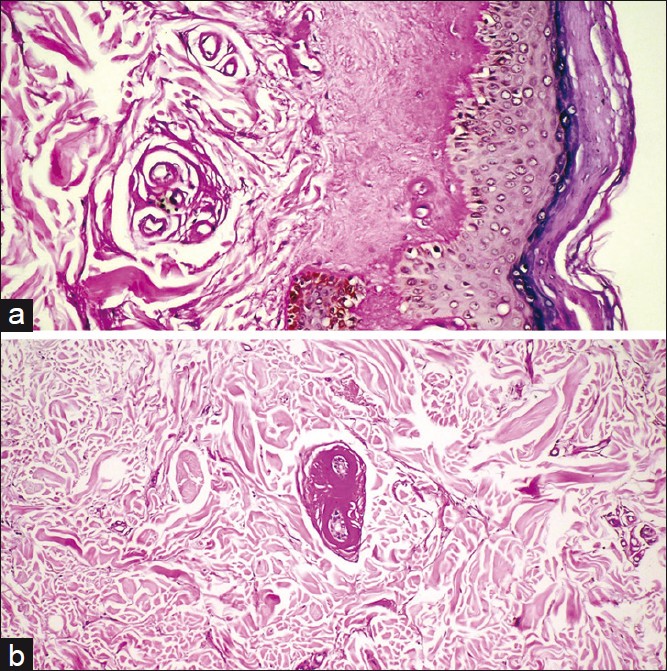 |
| Figure 2: Histopathology of the family 2 - patient. (a) Sheath - like hyaline material surrounding dermal capillaries (PAS, ×100). (b) Magnified picture of the same specimen (PAS, ×200) |
Family 3 (Patient 7-8)
As a coincidence, we came across a 31-year-old male patient. He came to our clinic together with his relative for hospital attendance. He had a prominent hoarseness of voice dating back to infancy. He was examined by otolaryngologist many times due to the hoarseness and its cause had not been identified. Family history revealed that his 33-year-old brother had similar lesions in milder character. Three other siblings were unaffected. Clinical examination of the affected individuals revealed hoarseness, multiple depressed scars on the face and extremities, beaded eyelid papules, and verrucous plaques on the knees and elbows [Figure - 3]. The younger patient′s frenulum was thickened, but movement was minimally affected. There was laryngeal thickening on indirect laryngoscopy.
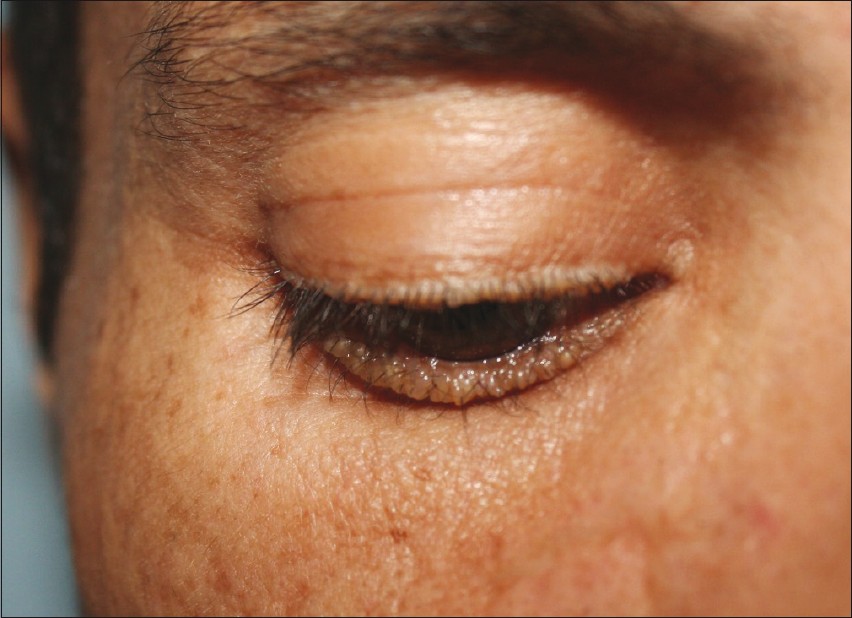 |
| Figure 3: Pearly eyelid papules on the family 3 - patient 7 |
Family 4 (Patient 9)
A 30-year-old woman presented with a complaint of itching and progressively thickening of the skin on her face and intertriginous areas. Her family history was unremarkable. Physical examination revealed beaded eyelid papules, mild verrucous plaques on the knee and elbow, acneiform scars, diffuse yellowish thickening on the face, and mild hoarseness of voice. In addition, brownish thick lesions were present in intertriginous areas which may be confused with acanthosis nigricans in the armpit. Histopathologic examination of the biopsy specimens taken from the eyelid and axilla revealed PAS-positive diffuse deposits of hyaline material. The axillary and eyelid lesions were diagnosed as LP. There was no history of seizures and her intelligence was normal, but she had dizziness. Cranial computed tomography (CT) revealed intracranial calcifications [Figure - 4].
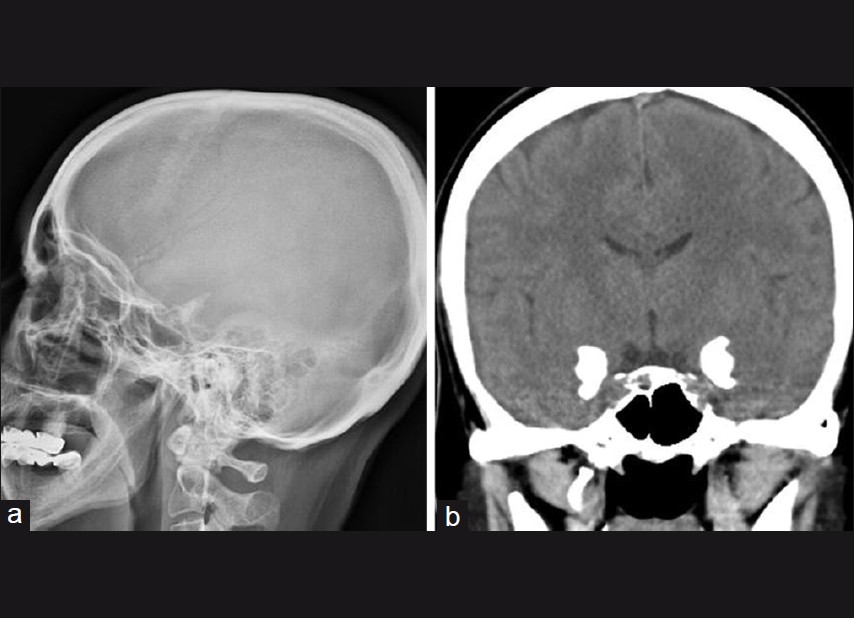 |
| Figure 4: (a) Temporal lobe calcification on direct cranial radiography. (b) Typical bilateral temporal lobe calcification was seen on cranial computed tomography, which is a diagnostic sign of lipoid proteinosis, on the family 4 - patient 9 |
Family 5 (Patient 10-12)
A 45-year-old woman complained of eyelid papules. Also, her two brothers (39- and 41-year-old) were affected. All three siblings had hoarseness of voice, beaded eyelid papules, and numerous waxy thickening over their face and extremities. The other six siblings were normal. The female patient′s tongue and frenulum were thickened, and she was unable to protrude the tongue out of her mouth [Figure - 5]. Indirect laryngoscopy showed laryngeal thickening in all affected siblings. Her cranial CT showed intracranial calcification, but there was no history of neurological symptoms.
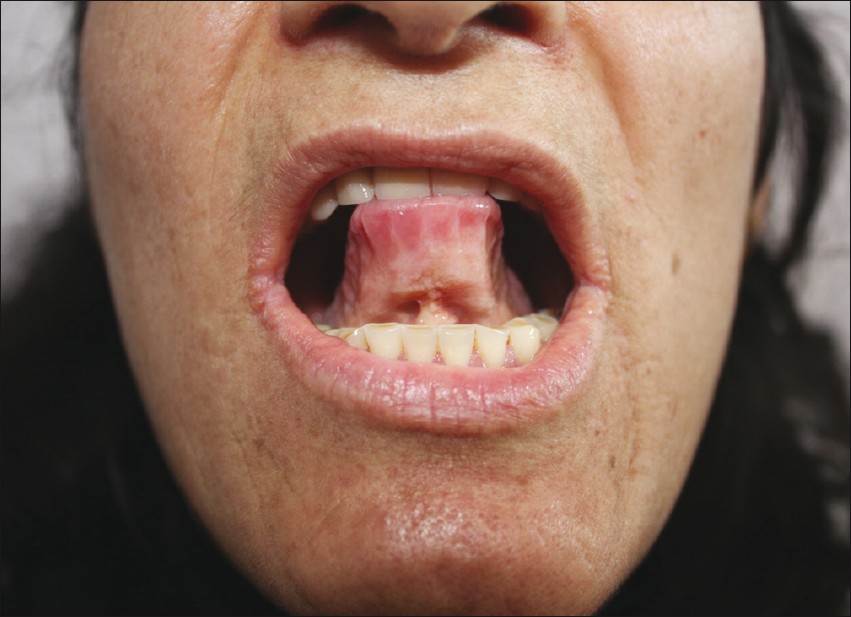 |
| Figure 5: Thickening of frenulum on the family 5 - patient 10 |
Family 6 (Patient 13-14)
A 17-year-old boy presented with hoarseness of voice and acneiform scars on his face and back. He had this complaint since he was a child. Family history revealed that his younger sister (16-year-old) had also similar symptoms. Two other siblings were unaffected.
Results
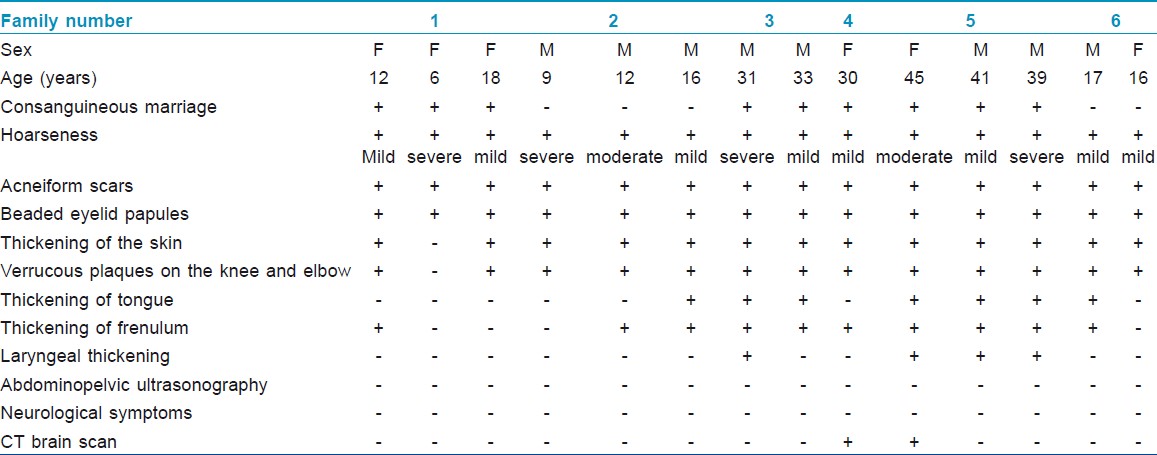
The clinical features of the 14 patients with LP are summarized in [Table - 1]. The 14 patients seen in our outpatient clinic were from six different families in Hatay, in the Eastern Mediterranean region of Turkey. All of the families, except two, had history of consanguineous marriage. All patients had typical cutaneous signs of LP and hoarseness of the voice. Two patients had intracranial calcifications. All patients′ abdominopelvic ultrasonographic examinations were normal. Severe laryngeal thickening was observed in four patients.
Discussion
LP is a rare disease worldwide. The disease is more common in the European and South African regions. In literature, the largest series of 29 LP patients have been reported from South Africa. Most of these patients were European descent, including South African descendants of German or Dutch immigrants. [6] Another report from northern Sweden described 27 patients belonging to 14 families. [7] As far as we know, there are 70 patients reported from Turkey. Of these reported cases, 14 were presented in European, Mediterranean and national congress, 16 were published in the Turkish literature, and 40 were published in the English literature. [8]
LP has recently been shown to result from loss of function mutations in the gene encoding extracellular matrix protein 1 (ECM1) on band 1q21. The most common location is being at exons 6 and 7 of ECM1. Patients with exon 6 mutation result in a more severe phenotype, but exon 7 mutations exhibit relatively milder clinical features. [9],[10] Loss of ECM1 may also have a role in skin changes including scarring, wound healing, and skin ageing. [9] ECM1 expression in human skin is regulated by age and ultraviolet exposure. Intrinsically aged skin shows a significantly reduced ECM1 expression in basal and upper epidermal cell layers compared with young skin. In photoaged skin, expression is significantly increased within the lower and upper epidermis. In LP, skin areas habitually exposed to the sun may show a more severely scarred and photoaged appearance. As the role of ECM1 in the ageing process of human skin is still largely unknown, [11] unfortunately, consanguineous marriages are common in the eastern part of the Turkey (range of 11.5-46%). [8] There is some evidence that the children of consanguineous marriage face a heightened risk of developing a range of genetic disorders. Because of this reason, we may suggest that autosomal recessive diseases such as LP are relatively more common in this region. In fact, all but two of our families had either a family history of a consanguineous marriage or a marriage of parents from the same village. All families were neither related to each other nor from the same ethnic group. Even though occurrence of siblings is considered to be rare in literature, in reality, many cases exist both in this study and in literature. [3],[4],[5] One of the most remarkable clinical features of LP is hoarseness, caused by infiltration of the vocal cords. This is usually the first symptom in the most that appears during infancy. The laryngeal changes may rarely be a cause of life-threatening symptoms as respiratory insufficiency. [3],[6],[7] Skin lesions may appear during the first two years of life and occur in two stages. The first consists of pustules, bullae, and hemorrhagic crusts of the skin, mouth, and throat. The skin lesions resolve with pock-like or acneiform scarring, usually on the face and distal extremities. In the second stage, deposits increase in the dermis, and the skin becomes thickened, yellowed, and waxy. Papules, plaques, and nodules take place primarily on the face but also in the axillae and on the scrotum. Beaded papules along the eyelid margins are characteristic findings. Verrucous lesions may occur on the extensor surfaces of the extremities, especially the elbow and the hands, usually after frictional trauma. Infiltration of the posterior tongue and frenulum may result in restricted mobility to the extent of causing speech difficulties. In addition to alopecia, dental anomalies and recurrent parotitis can occur. [2],[7],[8] All the patients were suffering hoarseness of voice and various types of skin lesions that include beaded eyelid papules, verrucous plaques on the knee and elbow, multiple depressed acneiform scars on the face and extremities. Moreover, in the past, an adult man was examined by otolaryngologist many times due to the hoarseness and its main cause had not been explained. One of the limitations of this report is that we only performed abdominopelvic ultrasonographic examination of to all patients for investigation of visceral involvement. There was no abnormality found according to this examination in all patients.
A pathognomonic finding of LP is bilateral intracranial bean-shaped calcifications within the temporal lobe or hippocampus-amygdala complex. CT may be helpful in the diagnosis. Neurologic manifestations may include epilepsy and neuropsychiatric abnormalities. [12] Two women patients studied herein had typical symmetrical temporal lobe calcifications observed on cranial CT. The neurological examinations of all patients were unremarkable.
LP is characterized histologically by disruption/duplication of basement membrane of vessels and widespread deposition of eosinophilic hyaline material in the dermis. This material is PAS-positive, but diastase-resistant. Deposits can also surround hair follicles, sebaceous glands, and rarely arrector pili muscles. In addition, dermal fibroblasts demonstrate characteristic cytoplasmic vacuole formation. [8]
LP has no effective treatment so far. Dermabrasion and chemical peeling applications are available for skin lesions, but these methods could provoke hyaline material deposition. Blepharoplasty and carbon dioxide laser surgery have improved beaded eyelid papules. Carbon dioxide laser surgery, microlaryngoscopy, and dissection of the vocal cords were performed in some cases . Several good clinical response were reported with oral dimethylsulfoxide, D-penicillamine, and retinoids. [13]
In conclusion, LP is relatively more common in Turkey. We found that previously reported 70 LP patients were from different regions in Turkey. We reported herein 14 additional cases that include siblings. Thus, the number of reported cases has reached 84. Compared with other countries in the world, most LP cases have been reported from Turkey. This unusual frequency suggests that this situation may be related to Turkey′s cross-continental location. It is important to note that this study is of significance because it concentrates on LP cases from a specific region. However, extensive further clinical and genetic studies are needed.
| 1. |
Urbach E, Wiethe C. Lipoidosis cutis et mucosae. Virchows Arch A Pathol Anat Histol 1929;27:286-319.
[Google Scholar]
|
| 2. |
Caplan RM. Visceral involvement in lipoid proteinosis. Arch Dermatol 1967;95:149-55.
[Google Scholar]
|
| 3. |
Nanda A, Alsaleh QA, Al-Sabah H, Ali AM, Anim JT. Lipoid proteinosis: Report of four siblings and brief review of the literature. Pediatr Dermatol 2001;18:21-6.
[Google Scholar]
|
| 4. |
Sethuraman G, Tejasvi T, Khaitan BK, Handa KK, Rao S, Singh MK, et al. Lipoid proteinosis in two siblings: A report from India. J Dermatol 2003;30:562-5.
[Google Scholar]
|
| 5. |
Nasiri S, Sarrafirad N, Kavand S, Saeedi M. Lipoid proteinosis: Report of three siblings. Dermatol Online J 2008;14:6.
[Google Scholar]
|
| 6. |
Van Hougenhouck-Tulleken W, Chan J, Hamada T, Thornton H, Jenkins T, McLean WH, et al. Clinical and molecular characterization of lipoid proteinosis in Namaqualand, South Africa. Br J Dermatol 2004;151:413-23.
[Google Scholar]
|
| 7. |
Hofer P. Urbach-Wiethe disease (lipoglycoproteinosis; lipoid proteinosis; hyalinosis cutis et mucosae): A clinico-genetic study of 14 families from northern Sweden. Hereditas 1974;77:209-18.
[Google Scholar]
|
| 8. |
Baykal C, Topkarci Z, Yazganoglu KD, Azizlerli G, Baykan B. Lipoid proteinosis: A case series from Istanbul. Int J Dermatol 2007;46:1011-6.
[Google Scholar]
|
| 9. |
Hamada T, Wessagowit V, South AP, Ashton GH, Chan I, Oyama N, et al. Extracellular matrix protein 1 gene (ECM1) mutations in lipoid proteinosis and genotype-phenotype correlation. J Invest Dermatol 2003;120:345-50.
[Google Scholar]
|
| 10. |
Chan I, Liu L, Hamada T, Sethuraman G, McGrath JA. The molecular basis of lipoid proteinosis: Mutations in extracellular matrix protein 1. Exp Dermatol 2007;16:881-90.
[Google Scholar]
|
| 11. |
Sander CS, Sercu S, Ziemer M, Hipler UC, Elsner P, Thiele JJ, et al. Expression of extracellular matrix protein 1 (ECM1) in human skin is decreased by age and increased upon ultraviolet exposure. Br J Dermatol 2006;154:218-24.
[Google Scholar]
|
| 12. |
Friedman L, Mathews RD, Swanepoel PD. Radiographic and computed tomographic findings in lipid proteinosis. A case report. S Afr Med J 1984;65:734-5.
[Google Scholar]
|
| 13. |
Rosenthal G, Lifshitz T, Monos T, Kachco L, Argov S. Carbon dioxide laser treatment for lipoid proteinosis (Urbach-Wiethe syndrome) involving the eyelids. Br J Ophthalmol 1997;81:253.
[Google Scholar]
|
Fulltext Views
2,345
PDF downloads
1,373





![[Figure - 1]](#fig_ijdvl_2012_78_3_318_95447_f2.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2012_78_3_318_95447_f3.jpg){kind=link}
![[Figure - 3]](#fig_ijdvl_2012_78_3_318_95447_f4.jpg){kind=link}
![[Figure - 4]](#fig_ijdvl_2012_78_3_318_95447_f5.jpg){kind=link}
![[Figure - 5]](#fig_ijdvl_2012_78_3_318_95447_f6.jpg){kind=link}
![[Table - 1]](#tbl_ijdvl_2012_78_3_318_95447_t1.jpg){kind=link}