Translate this page into:
Localized non-epidermolytic keratoderma
Correspondence Address:
R Chhetia
Department of Skin and VD, S.C.B. Medical College and Hospital, Cuttack, Orissa
India
| How to cite this article: Chhetia R, Jena D K, Dash M L, Ram M K. Localized non-epidermolytic keratoderma. Indian J Dermatol Venereol Leprol 2006;72:326 |
 |
|
 |
|
Sir,
Palmoplantar keratoderma is a heterogenous group of disorders, which encompass conditions characterizing thickening of palms and soles. It may be hereditary, acquired, syndromes with PPK as associated feature, diffuse or localized. Focal keratoderma is characterized by lesions located over pressure points. Focal NEPPK[1] or keratosis palmoplantaris varians or nummular PPK, is a hereditary transmitted condition with autosomal dominant inheritance. Clinical features usually appear in childhood and lesions are mainly located over the soles. Keratoderma may increase in severity in early adult life. There have been reports of intrafamilial and interfamilial variability, which accounts for the heterogenous nature of the disease. Palmar involvement is less common than plantar and can be nummular, linear and periungual, while plantar involvement is mostly over pressure points.
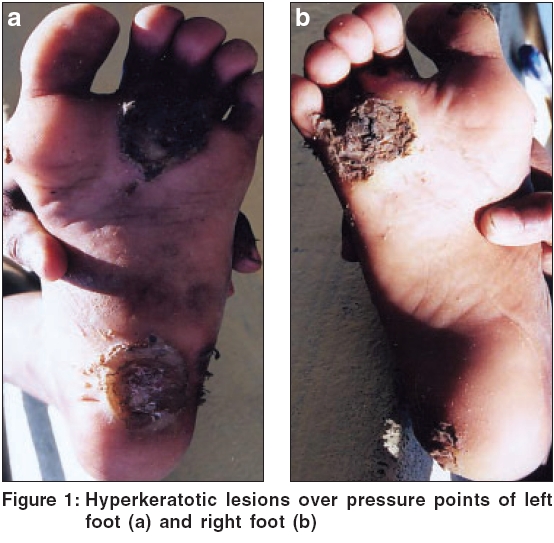
An eight year old boy presented with multiple painful, hyperkeratotic lesions over pressure points and bilateral soles. Keratoderma started at 5 years of age and was progressive since then. On examination, localized keratoderma over pressure points, bilateral soles and periungual distribution of great toe, was present [Figure - 1]. On paring, no bleeding points were observed. Lesions at presentation were secondarily infected. Follicular keratotic lesions over extremities were present. Oral cavity, nail and hair were normal.
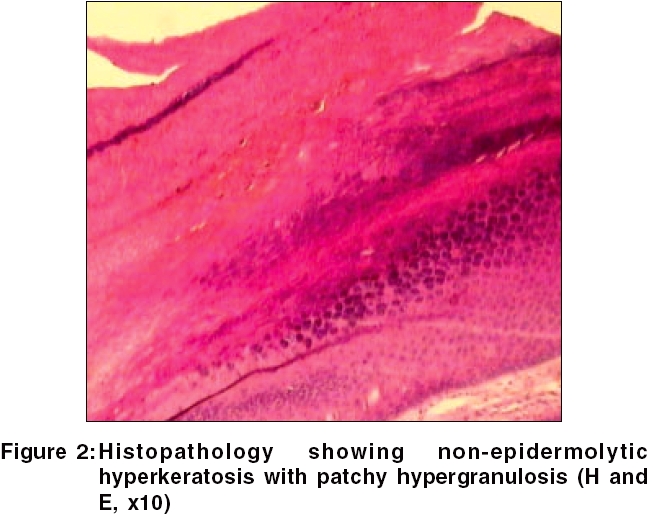
Similar types of lesions over soles were present in the grandfather of the child. Routine laboratory tests and VDRL were negative. Histopathology findings were suggestive of non-epidermolytic keratoderma [Figure - 2]. The patient was started on topical keratolytics and later on oral isotretinoin therapy. Only mild reduction in intensity of keratoderma was observed.
Focal NEPPK, often mentioned under painful hereditary callosities,[2],[3] is characterized by focal hyperkeratotic lesions, which develop at pressure bearing sites and sites of friction, usually of feet. Palm involvement depends on occupational exposure and is seen more in manual workers.[4] Keratoderma manifests in childhood and increases in severity in early adult life.[1] Focal NEPPK is associated with other clinical features like oral leukokeratosis, follicular lesions and some loss of axillary hair.[4] Linkage and haplotype analysis in NEPPK pedigree, suggested linkage to type I keratin cluster on chromosome 17. Point mutation in K-16 gene highly conserved domain (1A), is reported in two different pedigrees and K-16 mutations in both PC-1 and FNEPPK suggest both diseases to be variants of the same genetic disease.[5],[6]
The other focal keratodermas can be seen in oculocutaneous tyrosinemia, pachyonychia congenita, keratosis palmaris and plantaris with carcinoma of esophagus and focal epidermolytic PPK. Other conditions to be differentiated are viral warts, psoriasis, callosities, syphilis, yaws and leprosy. In our case, painful keratoderma over pressure points, family history and histopathology, confirmed the diagnosis of NEPPK.
The most challenging aspect of keratoderma is its treatment, as the options available provide only symptomatic and short term improvement. The various treatment options available include topical treatment such as keratolytics, paring, topical retinoids and topical (5% 5-Fluorouracil). Systemic treatment includes oral retinoids. Other options available are excision and grafting of hyperkeratotic skin.
In our patient, topical treatment provided little benefit. Oral isotretinoin 0.05-1 mg/kg provided only mild benefit to the patient and was withdrawn due to unbearable side effects. Finally, the patient resorted to topical keratolytics and paring. The challenge posed in the treatment of this inherited condition necessitates the search for more viable and permanent therapeutic options and application of gene therapy to treat the condition.
| 1. |
Lee R, Bowe WP, James WD, Guber PC, Ratnavel R. Keratosis palmaris et plantaris. In: Lebwohl MG, Wells MJ, Heymann WR, Quirk C, James WD. Editors. http://www.emedicine.com/DERM/topic589.htm, Last updated: March 23, 2006, Last accessed: April, 14, 2006.
[Google Scholar]
|
| 2. |
Roth W, Penneys NS, Fawcett N. Hereditary painful callosities. Arch Dermatol 1978;114:591-2.
[Google Scholar]
|
| 3. |
Wachters DH, Frensdorf EL, Hausman R, Van Dijk E. Keratosis palmoplantaris nummularis (hereditary painful callosities). J Am Acad Dermatol 1983;9:204-9.
[Google Scholar]
|
| 4. |
Griffiths WA, Judge MR, Leigh IM. Disorders of keratinization. In: Rook/ Wilkinson/ Ebling Text book of Dermatology. Champion RH, Burton JL, Burns DA, Breathnach SM, editors. 6th ed. Oxford: Blackwell; 1998. p. 1483.
[Google Scholar]
|
| 5. |
Kelsall DP, Stevens HP, Ratnavel R, Bryant SP, Bishop DT, Leigh IM, et al . Genetic linkage studies in non-epidermolytic palmoplantar keratoderma: Evidence for heterogeneity. Hum Mol Genet 1995;4:1021-5.
[Google Scholar]
|
| 6. |
Shamsher M, Navsaria HA, Stevens HP, Ratnavel RC, Purkis PE, Kelsall DP, et al . Novel mutation in keratin 16 gene underlie focal non-epidermolytic palmoplantar keratoderma in 2 families. J Invest Dermatol 1995;4:1875-81.
[Google Scholar]
|
Fulltext Views
2,377
PDF downloads
2,719





![[Figure - 1]](#fig_ijdvl_2006_72_4_326_26719_1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2006_72_4_326_26719_2.jpg){kind=link}