Translate this page into:
Maffucci syndrome in an eight-year-old girl
Correspondence Address:
Sharlene Helene H Chua
517 Camba Street, Binondo, Manila
Philippines
| How to cite this article: Chua SH, Frez MF. Maffucci syndrome in an eight-year-old girl. Indian J Dermatol Venereol Leprol 2015;81:412-414 |
Sir,
Maffucci syndrome is a rare, non-hereditary disorder characterized by multiple enchondromas and vascular malformations.
We report an 8-year-old girl with progressive multiple, bluish, compressible painless nodules confined to her right arm since birth [Figure - 1]. Older nodules had become doughy and hyperpigmented. There were no bruits or thrills. The right arm appeared larger than the left, however, measurement revealed no limb length discrepancy. Developmental milestones were appropriate for age and no similar symptoms were found in other family members. Physical examination did not reveal any other systemic involvement. Biopsy of a nodule revealed dilated vascular spaces in the dermis containing red blood cells and pink proteinaceous fluid, consistent with a hemo-lymphangioma [Figure - 2]. Radiograph revealed ill-defined lucent foci on the right hand, at the base of the fifth metacarpal, distal aspects of the proximal phalanges, distal aspects of the second to fifth middle phalanges, and second to fourth distal phalanges. These were interpreted as enchondromas. There were also punctate calcifications or phleboliths, on the right fifth metacarpo-phalangeal joint and distal fourth metacarpal [Figure - 3]. No fractures were noted. Ultrasound and magnetic resonance imaging (MRI) findings were also consistent with a hemangioma-lymphangioma complex. Complete blood count revealed no abnormality and fecal occult blood test was negative.
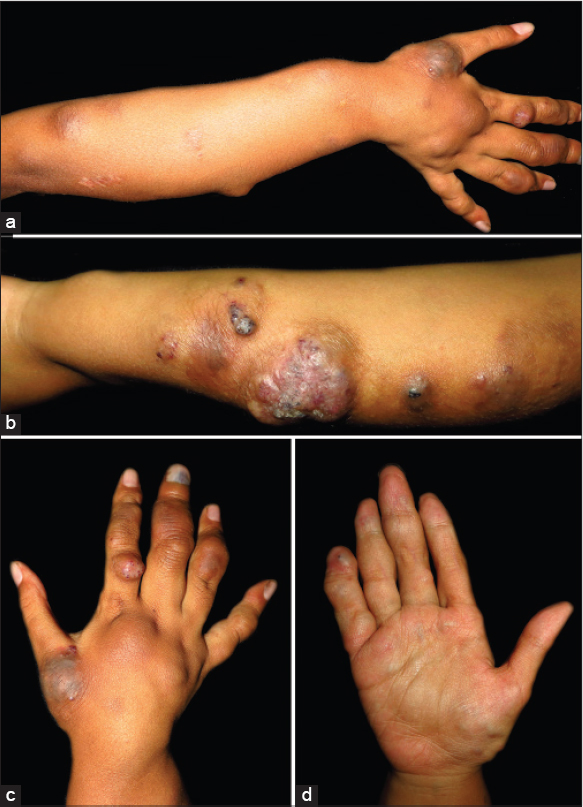 |
| Figure 1: Multiple bluish and hyperpigmented nodules on (a) dorsal aspect and (b) ulnar aspect of the right forearm and hand, (c) note the violaceous discoloration underneath the right third fingernail, (d) multiple compressible skin-colored and bluish nodules on right palm |
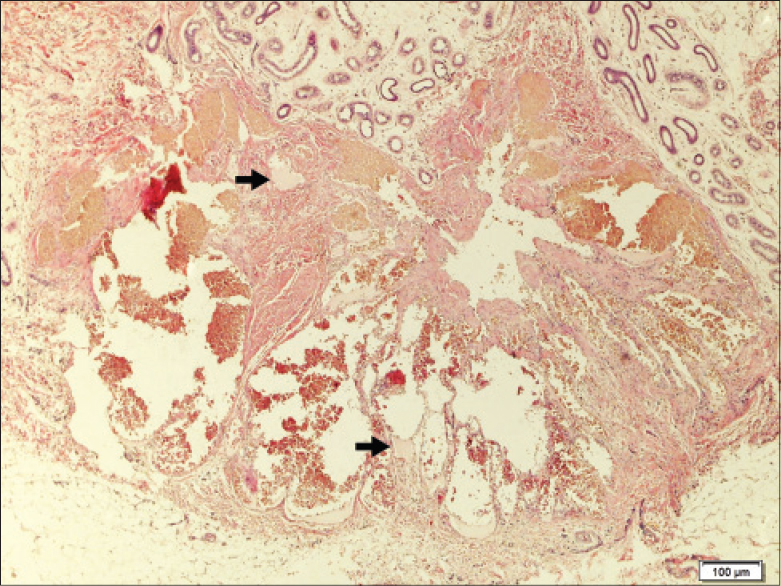 |
| Figure 2: Biopsy revealing collection of dilated vessels in the deep dermis lined by endothelial cells containing numerous erythrocytes and lymph (arrows) (H and E, X100) |
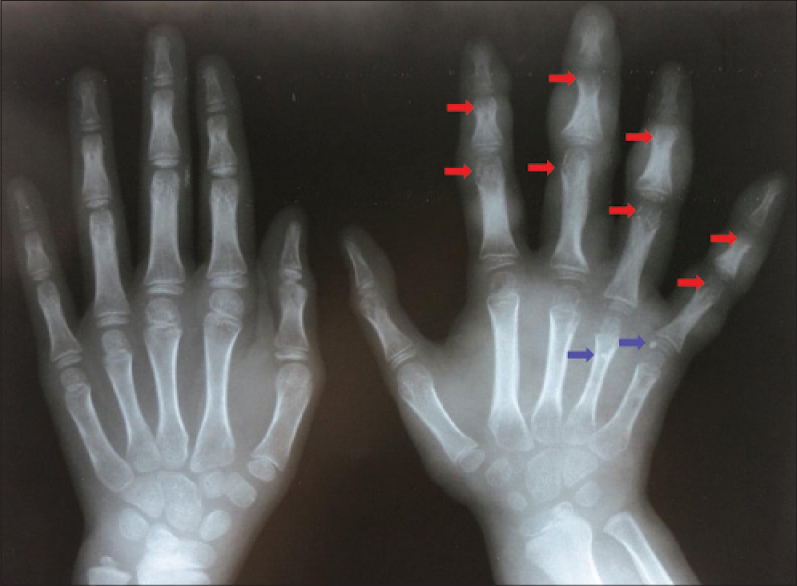 |
| Figure 3: Radiograph revealing early enchondroma formation (red arrows) and phleboliths (blue arrows) |
Differential diagnoses for a patient with unilateral limb involvement with vascular malformations would include Parkes-Weber Syndrome and Klippel-Trenaunay Syndrome. However, both disease entities present with capillary malformations manifested as cutaneous red stains and both would not have enchondromas as a feature. Furthermore, Parkes-Weber syndrome would present with bruits and thrills due to the underlying arterio-venous malformations. Glomuvenous malformations could also present as bluish nodules but these would not be compressible and the lesions would be painful. Blue rubber bleb nevus syndrome could also be one of the differential diagnoses as the syndrome can present with bluish, compressible venous malformations at birth and could progressively increase in number with age. However, unilateral involvement would be unusual in blue rubber bleb syndrome. Gastrointestinal involvement could present as anemia and gastrointestinal bleeding, both of which were not found in our patient. Ollier disease is the most common entity confused with Maffucci syndrome. However, in contrast to Maffucci syndrome, Ollier disease presents only with enchondromas and there would be no venous malformations.
The patient was co-managed by orthopedics, thoracovascular surgery and rehabilitation medicine. No invasive procedure was done because the lesions were asymptomatic and the procedures might do more harm with regard to function of her right arm. She was advised regular follow-up and monitoring to watch out for malignant transformation of lesions. Family counseling was done.
Maffucci syndrome was first described in 1881 by Angelo Maffucci. Only about 200 cases have been reported worldwide. [1] It has no racial or sexual predilection. Symptoms present before puberty in 78% of cases, before the age of 6 years in 45%, and at birth or within the first year of life in 25%. [2] Recently, it has been shown that somatic mosaic mutations in isocitrate dehydrogenase 1 (IDH1) and isocitrate dehydrogenase 2 (IDH2) underlie the tumorigenesis in Maffucci syndrome. [3]
Enchondromas are benign cartilaginous tumors usually found in the phalanges and the long bones. These tumors may also affect the tibia, fibula, humerus, ribs or cranium. Distribution is usually asymmetric. Enchondromas may be asymptomatic but pathological fractures may be found in 26% of cases. Risk of sarcomatous transformation ranges from 15% to 100% with chondrosarcoma being the most frequently associated. [2]
Vascular lesions may be venous malformations, spindle cell hemangiomas or sometimes, lymphangiomas. They are usually located adjacent to areas of enchondromatosis and present as bluish nodules. They may show a similar distribution with regard to laterality but may also be found in internal organs and mucous membranes. Malignant degeneration can occur. [4]
In the absence of symptoms or malignant transformation, no treatment is indicated. Regular monitoring should be done to ensure early detection of malignancies. Excision may be done. Osteotomy and curettage maybe done for bone lesions while sclerotherapy, irradiation, and laser therapy may be done for vascular lesions. [3],[4] Efficacy of rapamycin for refractory hemangioendotheliomas in Maffucci syndrome has also been recently reported. Mammalian target of rapamycin (mTOR) controls cell growth, proliferation, and survival. mTOR signaling is often upregulated in cancers and thus, there is growing interest in developing drugs that target this pathway. Rapamycin, an mTOR inhibitor, given 4 mg/day (2.5 mg/m 2 per day) to a 20-year-old male with Maffucci syndrome resulted in softening of nodules, decreased pain and less purplish lesions as early as 2 weeks into treatment. The disease was considered stable after 4 months of treatment and rapamycin was discontinued. At a follow-up of 16 months, lesions were stable in number and there was no recurrence of the pain. Adequate function of the hand was also preserved. [5]
| 1. |
Katz P, Colbert R, Drolet B. Unilateral mosaic cutaneous vascular lesions, enchondroma, multiple soft tissue chondromas and congenital fibrosarcoma - a variant of Maffucci syndrome? Pediatr Dermatol 2008;25:205-9.
[Google Scholar]
|
| 2. |
Flach HZ, Ginai AZ, Oosterhuis JW. Maffucci syndrome: Radiologic and pathologic findings. Radiographics 2001;21:1311-6.
[Google Scholar]
|
| 3. |
Amary MF, Damato S, Halai D, Eskandarpour M, Berisha F, Bonar F, et al. Ollier disease and Maffucci syndrome are caused by somatic mosaic mutations of IDH1 and IDH2. Nat Genet 2011;43:1262-5.
[Google Scholar]
|
| 4. |
Lissa FC, Argente JS, Antunes GN, Basso Fde O, Furtado J. Maffucci syndrome and soft tissue sarcoma: A case report. Int Semin Surg Oncol 2009;6:2.
[Google Scholar]
|
| 5. |
Riou S, Morelon E, Guibaud L, Chotel F, Dijoud F, Marec-Berard P. Efficacy of rapamycin for refractory hemangioendotheliomas for Maffucci′s syndrome. J Clin Oncol 2012;30:e213-5.
[Google Scholar]
|
Fulltext Views
3,777
PDF downloads
2,342





![[Figure - 1]](#fig_ijdvl_2015_81_4_412_157451_f1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2015_81_4_412_157451_f2.jpg){kind=link}
![[Figure - 3]](#fig_ijdvl_2015_81_4_412_157451_f3.jpg){kind=link}