Translate this page into:
McCune-Albright syndrome: A case report in a male
Correspondence Address:
Krina B Patel
Department of Dermatology, Smt. S.C.L. Hospital, Smt. NHL Municipal Medical College, Ahmedabad
India
| How to cite this article: Patel KB. McCune-Albright syndrome: A case report in a male. Indian J Dermatol Venereol Leprol 2010;76:723-724 |
Abstract
McCune-Albright syndrome (MAS) is a rare, heterogenous, clinical condition caused by a rare genetic mutation. The disorder is more common in females and is characterized by a triad of cutaneous, bone and endocrine abnormalities. We describe a male patient with MAS having multiple cafι-au-lait macules and deforming polyostotic fibrous dysplasia involving long bones of the limbs, skull and spine without any endocrine abnormality. Severe bone deformities involving almost all bones have not been described previously and this prompted us to present the current case.Introduction
McCune-Albright syndrome (MAS), also known as Albright syndrome, is a rare, heterogenous, clinical condition caused by a sporadic, somatic, post-zygotic mutation. The classical form of MAS is more common in females and is characterized by a triad of physical signs:
- Café-au-lait macules (CALMs) following lines of Blaschko
- Polyostotic fibrous dysplasia
- Autonomous, hyperfunctioning endocrinopathies
The non-classical form consists of only two of these three conditions. Gonadotropin-independent precocious puberty is the most common endocrine involvement described. A number of other endocrine and non-endocrine organs may be involved, including the adrenal, thyroid, pituitary, liver and heart.
Case Report
A 13-year old male patient with no history of consanguinity in the family presented with large patches of cutaneous pigmentation present since birth and multiple bony deformities developing for the last 5 years.
On examination, giant CALMs on the right side of the face, right upper back and buttock and the left upper back were observed [Figure - 1]a-c. CALMs were seen respecting the midline and had serrated margins (coast of maine) in comparison with smooth margins (coast of california) in neurofibromatosis.
The patient had gradually developing bony deformities of both lower limbs and right upper limb along with scoliosis and inability to walk without assistance. The patient had pain in the bones and had multiple pathological fractures occurring from time to time, which healed at a normal rate but with deformities. Extensive bowing deformity of both lower limbs and right upper limb was present [Figure - 2]a and b. The patient was under treatment of an orthopedic surgeon for repeated fractures of the long bones. Otherwise, the patient had normal developmental milestones and normal intelligence. He had just started to develop secondary sex characters in the form of pubic hair.
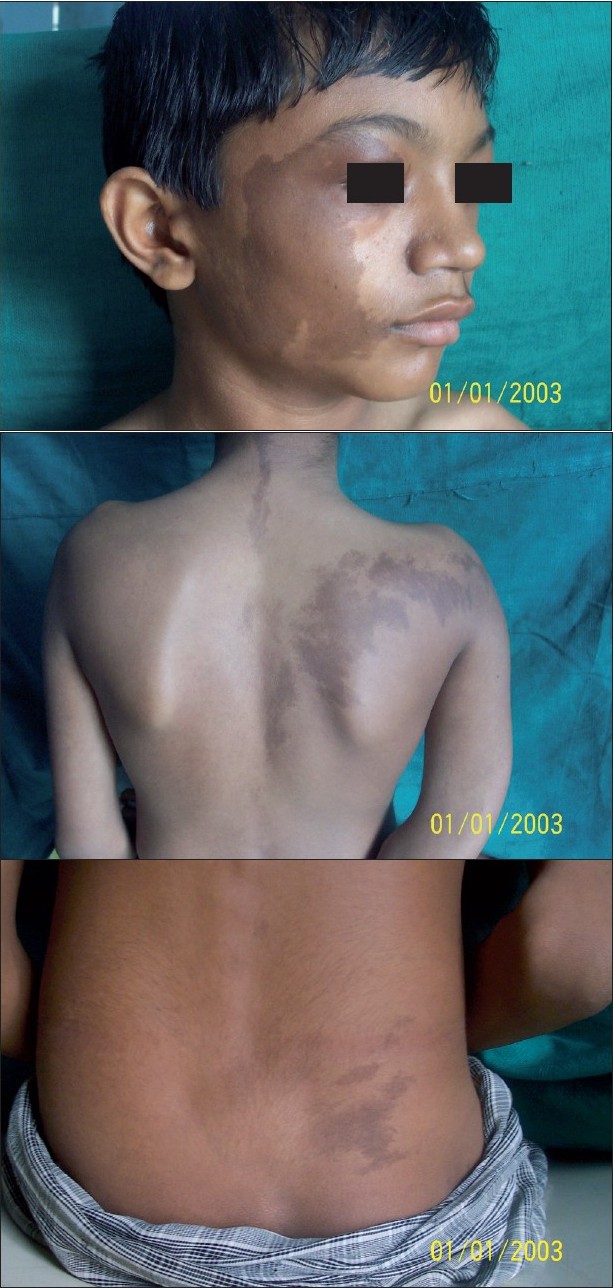 |
| Figure 1 :(a) Large cafe-au-lait macule with serrated margins on theright side of the face Figure 1b: Large cafe-au-lait macule on the right upper back and the small one on the left upper back respecting the midline Figure 1c: Cafe-au-lait macule on the right lower back |
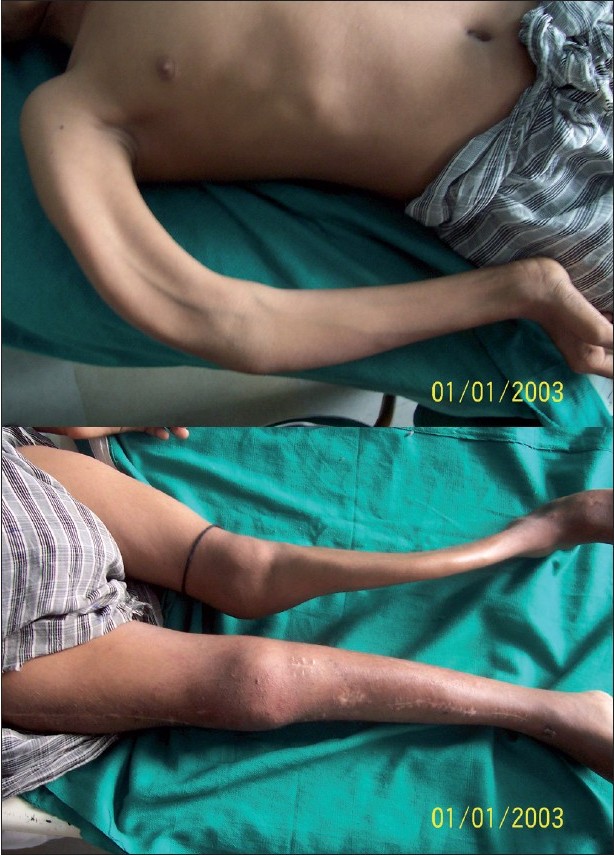 |
| Figure 2 :(a) Deformity of the right upper limb Figure 2b: Deformity of both lower limbs with scars of corrective orthopedic surgery on the right side |
X-rays of both the femur [Figure - 3]a and the tibia-fibula [Figure - 3]b showed extensive bowing deformities. Radiolucent and radiodense areas were observed in all affected long bones with replacement of marrow and loss of cortico-medullary differentiation. Right humerus and radius-ulna [Figure - 3]c showed similar changes and deformity. All the long bones showed evidence of healed old fractures. Involvement of femur bilaterally with progressive varus changes lead to the characteristic "Shepherd′s crook" deformity.
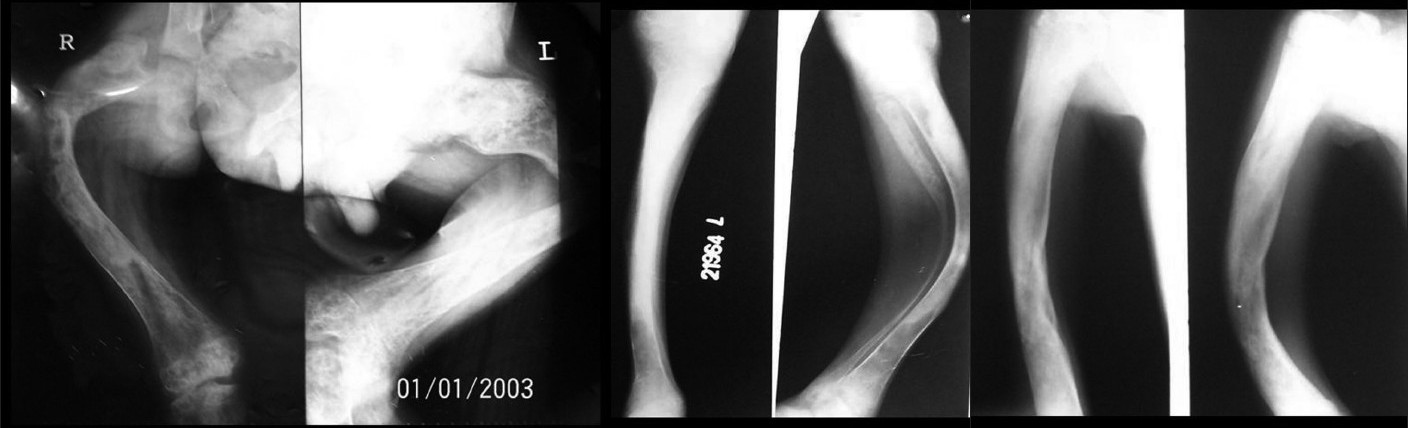 |
| Figure 3 :(a) Shepherd's crook deformity of both femurs on X-ray Figure 3b: Deformity of the tibia-fibula on X-ray Figure 3c: X-ray of the right humerus and radius-ulna showing deformity |
X-ray of the skull showed sclerosis of the base of the skull and widening of the diploae in the frontal and occipital region [Figure - 4]. Absence of frontal sinuses, sclerosis of the mandible and lesser wing of sphenoid were also evident. Ethmoid sinuses were replaced by sclerotic bone. X-ray of the spine showed fibrous dysplasia of the vertebrae and scoliotic deformity.
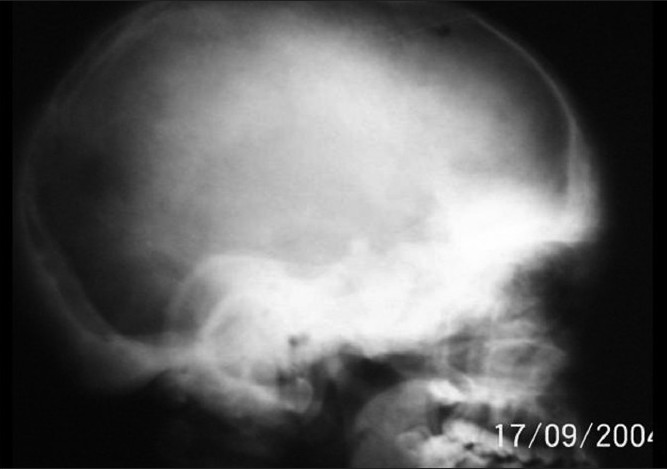 |
| Figure 4 :X-ray of the skull showing basal sclerosis |
Bone biopsy showed narrow, curved and irregular-shaped trabeculae having the characteristic "fish-hook" configuration interspersed with fibrous tissue. The entire appearance was that of "Chinese letter" or "alphabet soup" [Figure - 5].
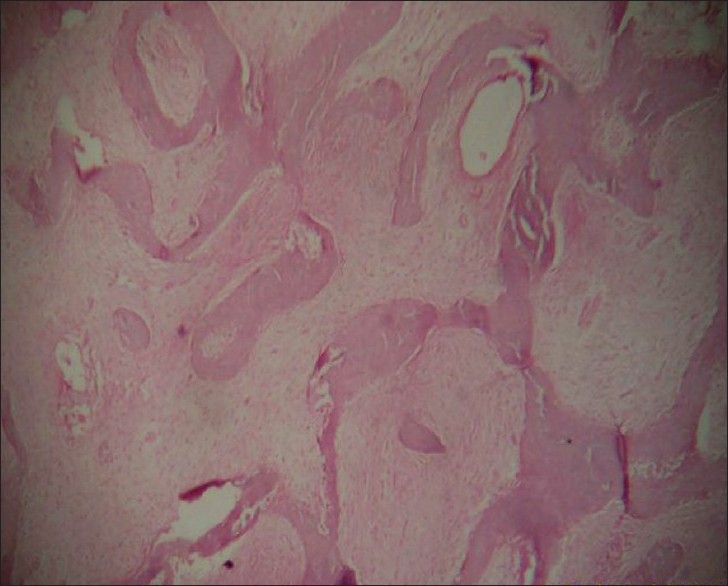 |
| Figure 5 :Histopathology of the bone showing the fish-hook appearance (H and E, ×40) |
Biochemical investigations showed serum calcium within the reference range but markedly increased level of serum alkaline phosphatase: 541 mg/dl (reference range: 25-145 mg/dl). Thyroid function tests were within the reference range.
As, clinically, no other endocrine abnormality was evident, other endocrine investigations were not carried out.
All these interesting clinical, radiological, biochemical and biopsy findings confirm the diagnosis of MAS.
Discussion
MAS is a rare multisystem disorder, first described in 1937 separately by Donovan McCune and Fuller Albright in a group of children, mostly females, with skin pigmentation, bone deformities and endocrinal disorders. [1]
The disorder is the result of post-zygotic somatic mutation in the gene GNAS 1 on chromosome 20q13-13.2 9, [2] coding for the alpha subunit of stimulatory G protein (Gsa).
G proteins couple cell surface receptors to intracellular proteins to activate or inactivate signaling cascades. The stimulatory G protein is normally activated when a hormone or other ligand binds to the cell surface receptor. The activated Gsa subunit subsequently dissociates from the receptor, binds to adenylyl cyclase and stimulates an increase in the intracellular cyclic adenosine monophosphate (cAMP) levels. The Gsa subunit is then inactivated, which re-associates with the receptor and is again available for hormone-mediated reactivation.
The specific mutation that causes MAS occurs at a site in the protein that mediates the inactivation of the Gsa subunit. Once activated, the mutated Gs alpha subunit remains activated for a prolonged period despite the absence of hormone stimulation of the receptor. The result is constitutive activation of the Gsa subunit, constant stimulation of adenylyl cyclase and persistently high levels of intracellular cAMP. In various tissues, increased cAMP levels can mediate mitogenesis and increased cell function. The specific phenotype depends on the cell type containing the mutation. [3],[4]
The clinical expression depends on the number of mutated cells and affected organs. Thus, the presentation can be heterogenous, involving various endocrine and non-endocrine organs. It can be early or late onset or slow or quick evolution.
Because MAS results from post-zygotic somatic mutation, all the daughter cells of the embryonic cell in which the initial mutation occurred also contain the mutation. The earlier the mutation occurs in embryogenesis, the more widespread are the tissues involved. Mutations late in embryogenesis are more focused and account for mild cases, with only one or two of the three phenotypic features of the syndrome.
As this mutation is lethal in the germ cell line, autosomal transmission is not seen, although the disorder has been reported in monozygotic twins.
The clinical presentation of MAS is highly variable, depending on which of the various potential components of the syndrome predominate. The classical form of MAS is more common in females and is defined by a triad of physical signs as described above.
Gonadotropin-independent precocious puberty is the most common endocrine manifestation. Other endocrine organ involvement may lead to hyperthyroidism, hypercortisolism, growth hormone excess, hyperprolactinemia, etc. Hepatobilliary disorders, hyperphosphatemia, intestinal polyposis, cardiac involvement, etc. are non-endocrine manifestations. [5]
CALMs develop between the age of 4 months and 2 years, but may be present at birth. Extensive light brown patches with serrated margins occur mainly on the trunk, buttocks and thighs. They are asymmetrical and most extensive on the side showing the most severe bone involvement. The unique pattern of CALMs is that they never cross the midline. Histopathology shows extensive basal layer pigmentation and, occasionally, giant melanosomes, but is not diagnostic.
Bone dysplasia is revealed during the first decade by aching pain, pathological fractures, limb asymmetry and deformities. Abnormal fibrous tissue growth occurs in many bones, especially in the long bones, ribs and skull bones. Fibrous dysplasia ranges from small asymptomatic areas, detectable only by bone scan, to markedly disfiguring lesions, resulting in pathological fractures and impingement on vital structures. Radiographic appearance can be pagetoid (56%), sclerotic (23%) or cystic (21%). [6] Although once considered a rare finding, fibrous dysplasia of the vertebrae leading to scoliosis is found in 40% of the patients with polyostotic fibrous dysplasia in a study by Leet et al. [7]
Histopathology of the affected bone shows narrow, curved and misshaped bone trabeculae, often having a characteristic "fish-hook" configuration, interspersed with fibrous tissue of variable cellularity. Ultrastucturally, the immature woven bone trabeculae are lined by abnormal osteoblasts with fibroblast-like appearance. [8]
Depending on other organ involvement, biochemical investigations show elevated estradiol and suppressed gonadotropin levels, abnormal liver or thyroid function tests, hypophosphatemia, etc.
No definite treatment is available for MAS and, to date, prenatal diagnosis is not possible. But, recently, through novel polymerase chain reaction-based techniques, activating mutation in the peripheral blood of patients with MAS has been successfully detected, which might help in diagnostic as well therapeutic areas. [2],[9]
Various medical and surgical treatments are offered for endocrine and non-endocrine involvement according to signs and symptoms.
Corrective surgery can be performed for bone dysplasia. Recently, bisphosphonate therapy has shown promise for fibrous dysplasia patients as it helps to diminish pain, prevents fracture and leads to partial resolution of fibrous dysplasia lesions. [10]
MAS has been reported in Indian studies, [11],[12] but most of the reported cases in the literature are female and cases with endocrine abnormalities as the prominent feature. We report MAS in a male patient with some interesting findings, like:
- Facial CALM, which is rarely seen
- Severe bone deformities of the long bones as well as scoliosis, making the patient handicapped
- Involvement of the non-weight bearing bones also
- Bilateral bone involvement is rarely reported in the literature, which is also an extremely rare finding seen in our patient, although it followed the rule of more severe bone involvement on the side of the body having more extensive CALMs (right side in our patient).
| 1. |
Albright F, Butler AM, Hampton AO. Syndrome characterized by osteitis fibrosa disseminata, areas of pigmentation and endocrine dysfunction, with precocious puberty in females. N Engl J Med 1937;216:727-47.
[Google Scholar]
|
| 2. |
DiCaprio MR, Enneking WF. Fibrous dysplasia. Pathophysiology, evaluation, and treatment. J Bone Joint Surg Am 2005;87:1848-64.
[Google Scholar]
|
| 3. |
Spiegel AM. The molecular basis of disorders caused by defects in G proteins. Horm Res 1997;47:89-96.
[Google Scholar]
|
| 4. |
Weinstein LS, Shenker A, Gejman PV, Merino MJ, Friedman E, Spiegel AM. Activating mutations of the stimulatory G protein in the McCune-Albright syndrome. N Engl J Med 1991;325:1688-95.
[Google Scholar]
|
| 5. |
Shenker A, Weinstein LS, Moran A, Pescovitz OH, Charest NJ, Boney CM, et al. Severe endocrine and nonendocrine manifestations of the McCune-Albright syndrome associated with activating mutations of stimulatory G protein GS. J Pediatr 1993;123:509-18.
[Google Scholar]
|
| 6. |
Singer FR. Fibrous dysplasia of bone: the bone lesion unmasked. Am J Pathol 1997;151:1511-5.
[Google Scholar]
|
| 7. |
Leet AI, Magur E, Lee JS, Wientroub S, Robey PG, Collins MT. Fibrous dysplasia in the spine: Prevalence of lesions and associations with scoliosis. J Bone Joint Surg Am 2004;86:531-7.
[Google Scholar]
|
| 8. |
Rosai J. Bone and joints. In: Rosai J, editor. Ackerman's Surgical Pathology. 8 th ed. St. Louis: Mosby; 1996. p. 1981-2.
th ed. St. Louis: Mosby; 1996. p. 1981-2.'>[Google Scholar]
|
| 9. |
Lietman SA, Ding C, Levine MA. A highly sensitive polymerase chain reaction method detects activating mutations of the GNAS gene in peripheral blood cells in McCune-Albright syndrome or isolated fibrous dysplasia. J Bone Joint Surg Am 2005;87:2489-94.
[Google Scholar]
|
| 10. |
Lane JM, Khan SN, O'Connor WJ, Nydick M, Hommen JP, Schneider R, et al. Bisphosphonate therapy in fibrous dysplasia. Clin Orthop Relat Res 2001;382:6-12.
et al. Bisphosphonate therapy in fibrous dysplasia. Clin Orthop Relat Res 2001;382:6-12.'>[Google Scholar]
|
| 11. |
Rao S, Colaco MP, Desai MP. McCune Albright syndrome (MCAS): A case series. Indian Pediatr 2003;40:29-35.
[Google Scholar]
|
| 12. |
Kumawat DC, Bomb BS, Agarwal HK, Bhatnagar HN. The Albright syndrome associated with acromegaly and hyperthyroidism (a case report). J Postgrad Med 1987;33:155-7.
[Google Scholar]
|
Fulltext Views
5,486
PDF downloads
1,653





![[Figure - 1]](#fig_ijdvl_2010_76_6_723_72473_f1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2010_76_6_723_72473_f2.jpg){kind=link}
![[Figure - 3]](#fig_ijdvl_2010_76_6_723_72473_f3.jpg){kind=link}
![[Figure - 4]](#fig_ijdvl_2010_76_6_723_72473_f4.jpg){kind=link}
![[Figure - 5]](#fig_ijdvl_2010_76_6_723_72473_f5.jpg){kind=link}