Translate this page into:
Novel and recurrent mutations in GJB3 and GJB4 cause erythrokeratodermia variabilis et progressiva
2 Department of Dermatology, Peking University First Hospital; Beijing Key Laboratory of Molecular Diagnosis on Dermatoses; Peking-Tsinghua Center for Life Sciences; Academy for Advanced Interdisciplinary Studies, Peking University, Beijing, China
Correspondence Address:
Zhimiao Lin
Department of Dermatology, Peking University First Hospital, Beijing 100034
China
| How to cite this article: Dai S, Wang H, Lin Z. Novel and recurrent mutations in GJB3 and GJB4 cause erythrokeratodermia variabilis et progressiva. Indian J Dermatol Venereol Leprol 2020;86:87-90 |
Sir,
Erythrokeratodermia variabilis et progressiva comprises a group of genetically heterogeneous skin disorders characterized by the coexistence of erythematous patches and persistent hyperkeratotic plaques. It can be caused by mutations in GJB3, GJB4, GJA1, KDSR, and KRT83, encoding connexin 31, connexin 30.3, connexin 43, 3-ketodihydrosphingosine reductase, and keratin 83, respectively.[1],[2],[3] We herein report three unrelated families with this condition harboring one recurrent mutation in GJB3 and two novel mutations in GJB4.
Three families of Chinese Han ethnicity were referred to the Peking University First Hospital in Beijing, China. They were clinically diagnosed to have erythrokeratodermia variabilis et progressiva [Figure - 1]. The age of onset ranged from at birth to adolescence. All the affected individuals exhibited persistent hyperkeratotic plaques as well as transient symmetric red-brown erythematous patches which lasted hours to months. Two probands had relatively restricted lesions which predominated in body folds (proband 1) [Figure - 2]a and dorsal feet (proband 3) [Figure - 2]b, whereas proband 2 showed progressive lesions extending over the limbs and trunk with mild skin peeling and scaling [Figure - 2]c and d]. Proband 2 had palmoplantar keratosis also. Seasonal changes and emotional stress caused exacerbation of the lesions.
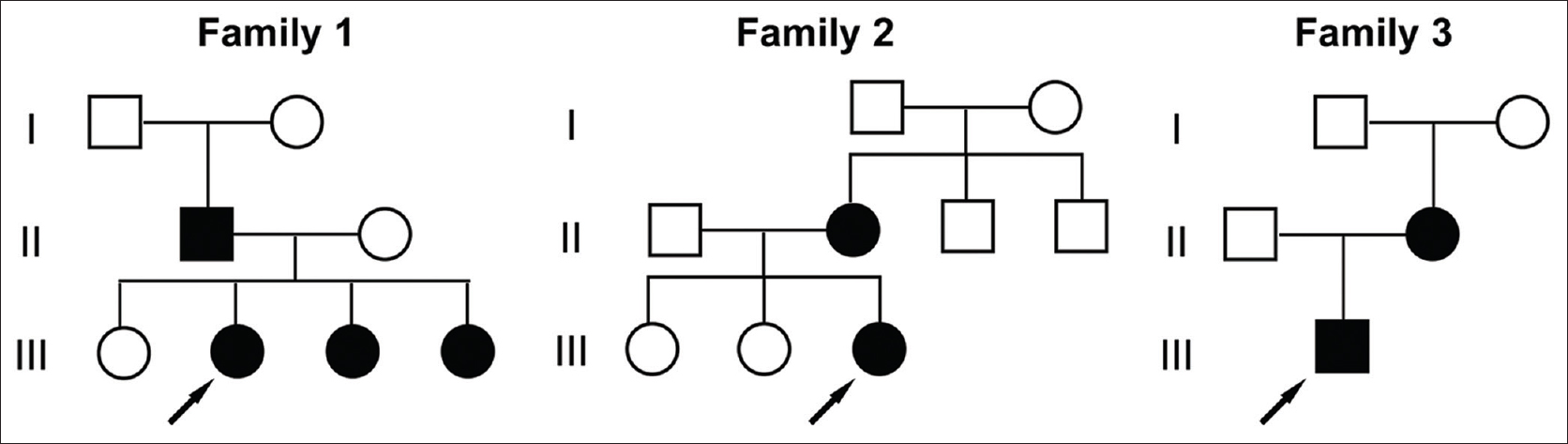 |
| Figure 1: Pedigrees of the three families with erythrokeratodermia variabilis et progressiva |
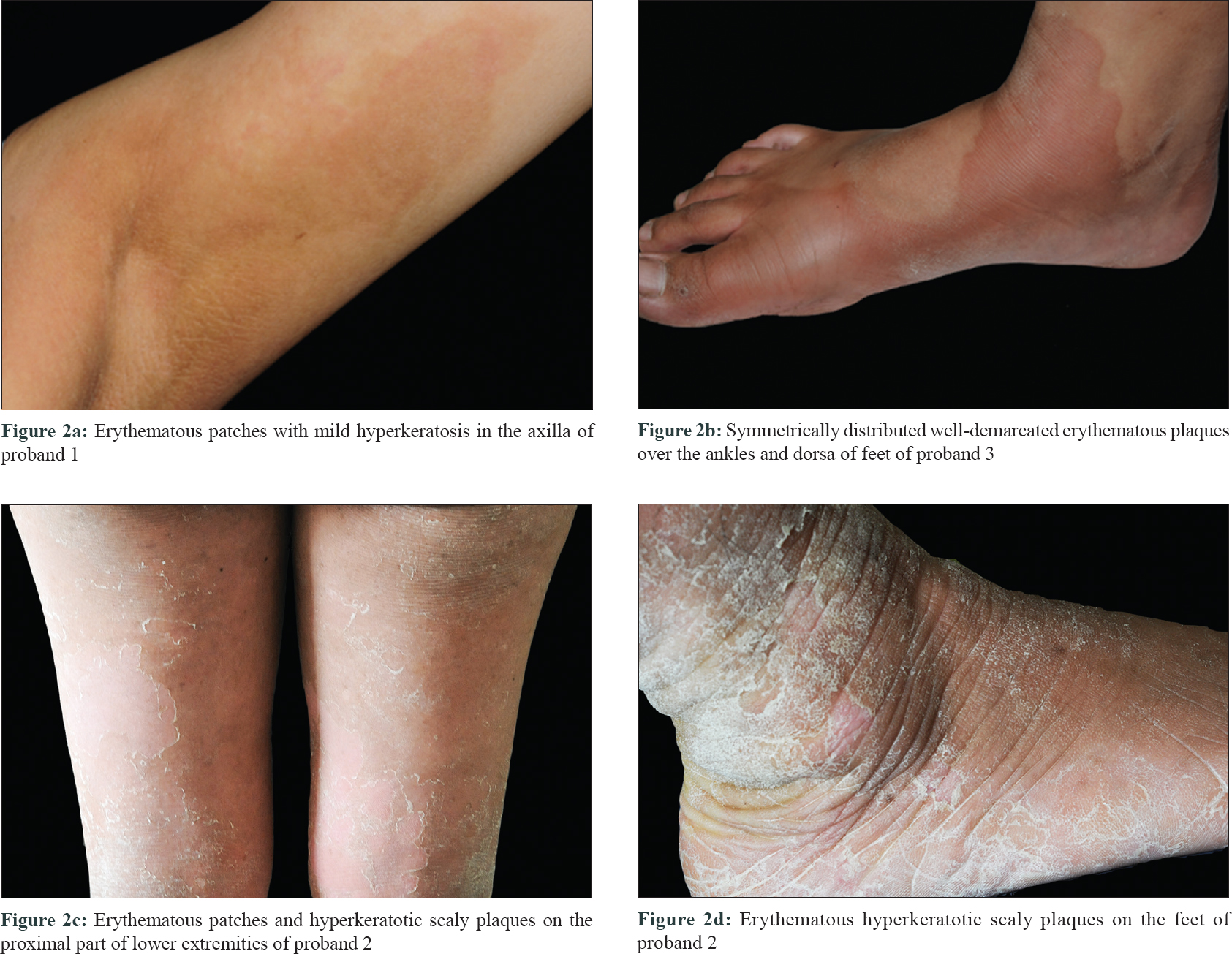 |
| Figure 2: |
After obtaining written informed consent for genetic testing from the probands and the approval from institutional ethics committee of Peking University First Hospital, a screening was done for exons and their flanking regions of GJB3 and GJB4, the most common causative genes of erythrokeratodermia variabilis et progressiva, in all the probands. Sanger sequencing revealed two previously unreported heterozygous mutations, c.77C>A (p. Ser26Tyr) and c.182C>G (p. Pro61Arg) in GJB4, in the first and second proband respectively and one recurrent heterozygous mutation c.256T>A (p. Cys86Ser) was seen in GJB3 in the third proband. [Figure - 3]a. Co-segregation of the mutations with phenotype was confirmed in all three families. The two novel missense mutations, c.77C>A and c.182C>G in GJB4, were absent in all public databases including ExAC, gnomAD, and 1000G and were predicted to be “disease causing” by MutationTaster.
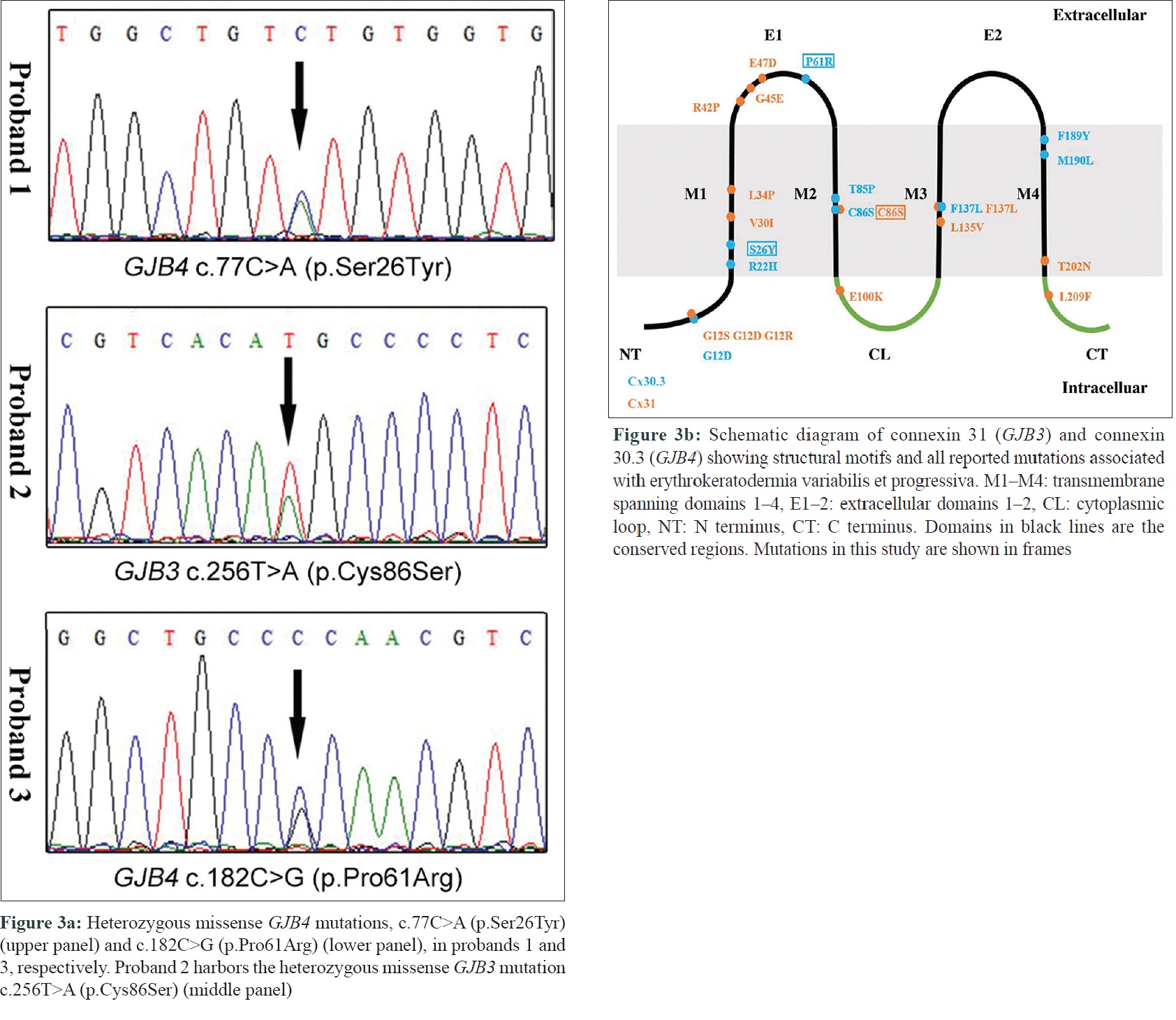 |
| Figure 3: |
Gap junction channels, which mediate direct intercellular signaling, play important role in many biological processes including development, differentiation, and cell synchronization. Gap junction channel is structured by two hemichannels docking end-to-end, each formed by six connexins oligomerized together.[4] Connexin 31 and 30.3, encoded by GJB3 and GJB4, respectively, are members of the highly homologous connexin protein family, which share a common structure topology characterized by four transmembrane domains (M1–M4) linked by two extracellular loop (E1 and E2) and one intracellular loop [Figure - 3]b. The transmembrane and extracellular domains along with the N-terminus are defined as the conservative domains. To date, we have found more than 30 previous reports of mutations in GJB3 and GJB4 to be associated with erythrokeratodermia variabilis et progressiva, including the two novel mutations reported here. Most of these mutations occurred in the conservative regions [Figure - 3]b. Nevertheless, none of the previously reported GJB4 mutations occurred in extracellular domains, which function as essential element in the formation of the gap junction pore.[5] Among all previously reported mutations associated with erythrokeratodermia variabilis et progressiva, the novel mutation c.182C>G (p. Pro61Arg) reported here is the first to be located in the first extracellular (E1) domain of GJB4 and also the first in the extracellular domain of the protein. The other novel mutation of GJB4 (p. Ser26Tyr) is located in the transmembrane region, disruption of which is likely to interfere in channel gating.[5] The GJB3 mutation (p. Cys86Ser) in proband 2 was repeatedly described elsewhere, and the severity of the condition varied greatly.[1] Proband 2 is the most severely affected, showing generalized erythema and hyperkeratosis, combined with other manifestations such as palmoplantar keratosis, scaling, and ridging in large skin folds. This corroborates the fact of great interfamilial phenotypic heterogeneity in erythrokeratodermia variabilis et progressiva. Although the general clinical presentation of patients with GJB4 mutations was similar to those with GJB3 mutations, previous studies have suggested certain correlation between genotype and phenotype of this disorder.[5] It has been reported that patients harboring the mutations, p. Gly12Asp and p. Phe137Leu in GJB4, seemed to manifest milder diseases than those with the corresponding mutations in GJB3.[5] By reviewing and comparing the phenotype of patients having mutations in GJB3 and GJB4 affecting homologous residues in previous studies and our study it can be understood that those with mutations in GJB3 tend to have more severe phenotype than those with mutations in GJB4. Hence we recommend a prior screening of GJB3 in patients with generalized disease and palmoplantar keratosis in genetic counseling for erythrokeratodermia variabilis et progressiva in future. However, factors including age and previous treatment which might affect the clinical severity were not taken into account while comparing the phenotype of patients with GJB3 and GJB4 mutations. So further functional studies of the mutations are required to confirm this observation.
On the whole this study expands the mutation spectrum of GJB3 and GJB4 and further suggests a possible genotype–phenotype correlation in erythrokeratodermia variabilis et progressiva.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patients have given their consent for their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
National Natural Science Foundation of China (grant nos. 81573032 and 81872515) and China National Funds for Excellent Young Scientists (grant no. 81522037).
Conflicts of interest
There are no conflicts of interest.
| 1. |
Richard G, Smith LE, Bailey RA, Itin P, Hohl D, Epstein EH Jr., et al. Mutations in the human connexin gene GJB3 cause erythrokeratodermia variabilis. Nat Genet 1998;20:366-9.
[Google Scholar]
|
| 2. |
Macari F, Landau M, Cousin P, Mevorah B, Brenner S, Panizzon R, et al. Mutation in the gene for connexin 30.3 in a family with erythrokeratodermia variabilis. Am J Hum Genet 2000;67:1296-301.
[Google Scholar]
|
| 3. |
Boyden LM, Craiglow BG, Zhou J, Hu R, Loring EC, Morel KD, et al. Dominant de novo mutations in GJA1 cause erythrokeratodermia variabilis et progressiva, without features of oculodentodigital dysplasia. J Invest Dermatol 2015;135:1540-7.
[Google Scholar]
|
| 4. |
Bai D, Wang AH. Extracellular domains play different roles in gap junction formation and docking compatibility. Biochem J 2014;458:1-0.
[Google Scholar]
|
| 5. |
Richard G, Brown N, Rouan F, Van der Schroeff JG, Bijlsma E, Eichenfield LF, et al. Genetic heterogeneity in erythrokeratodermia variabilis: Novel mutations in the connexin gene GJB4 (Cx30.3) and genotype-phenotype correlations. J Invest Dermatol 2003;120:601-9.
[Google Scholar]
|
Fulltext Views
4,882
PDF downloads
2,892





![[Figure - 1]](#fig_ijdvl_2020_86_1_87_272124_f1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2020_86_1_87_272124_f2.jpg){kind=link}
![[Figure - 3]](#fig_ijdvl_2020_86_1_87_272124_f3.jpg){kind=link}