Translate this page into:
Oculocutaneous tyrosinemia: A case report with delayed diagnosis and excellent response to dietary modification
2 Department of Pediatrics, Division of Metabolism, Istanbul University Cerrahpasa Medical Faculty, Istanbul, Turkey
3 Institute of Human Genetics, University of Bonn, Bonn, Germany
4 Department of Ophthalmology, Marmara University School of Medicine, Istanbul, Turkey
Correspondence Address:
Burak Tekin
Marmara University Pendik Training and Research Hospital Department of Dermatology, Mimar Sinan Street, No: 41. Pendik/Istanbul
Turkey
| How to cite this article: Tekin B, Yucelten D, Zeybek CA, Kiykim E, Wehner M, Betz RC, Toker AE. Oculocutaneous tyrosinemia: A case report with delayed diagnosis and excellent response to dietary modification. Indian J Dermatol Venereol Leprol 2015;81:303-305 |
Sir,
Oculocutaneous tyrosinemia (tyrosinemia type II, Richner-Hanhart syndrome) is a rare inherited disorder involving tyrosine metabolism. Ocular symptoms classically appear months to years before the typically painful palmoplantar hyperkeratosis. Although many of the reported families are known to be of Italian origin, [1] the disease has also been reported from other areas of the world, including India. [2]
An 11-year-old girl, born to a consanguineous marriage, presented with painful plantar hyperkeratosis that she had suffered, from the age of 2 years. She was admitted to the emergency department several times during her childhood due to severe attacks of pain that affected walking. A punch biopsy from the sole of her foot taken 5 years ago revealed compact orthohyperkeratosis, hypergranulosis, and epidermal hyperplasia consistent with palmoplantar keratoderma. She had ocular surgery to treat bilateral corneal ulcers when she was aged 1 year, with a presumptive diagnosis of herpes. Dermatological examination revealed sharply demarcated, yellowish hyperkeratotic plaques localized to pressure bearing areas on both plantar surfaces and the palmar surface of her fingertips [Figure - 1]a and [Figure - 2]a. The lesions were tender to palpation and accompanied by hyperhidrosis. Bilateral irregular astigmatism due to corneal opacities was noted on eye examination. Her maternal uncle had similar plantar lesions and her maternal grandmother experienced early deterioration of vision. Her parents and 7-year-old sister did not have any skin or eye complaints.
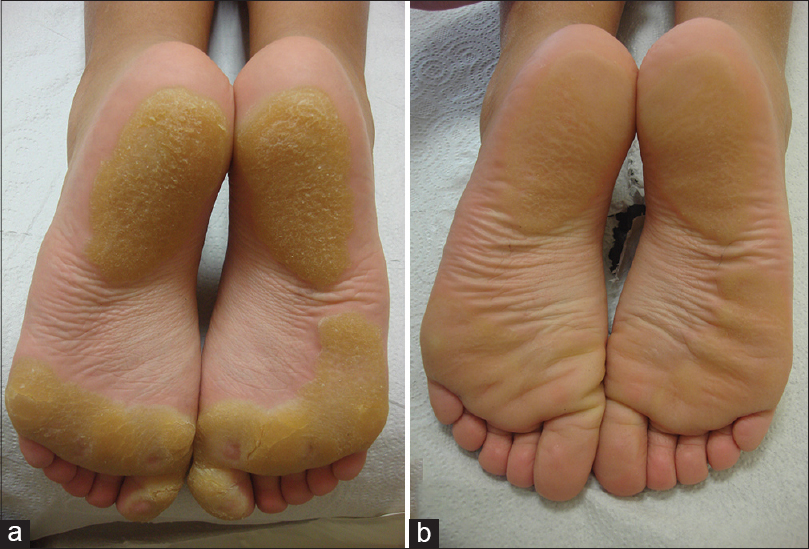 |
| Figure 1: Yellowish focal hyperkeratotic plaques without an erythematous border on the soles (a) before, and (b) 3 weeks after the patient started a tyrosine-restricted diet |
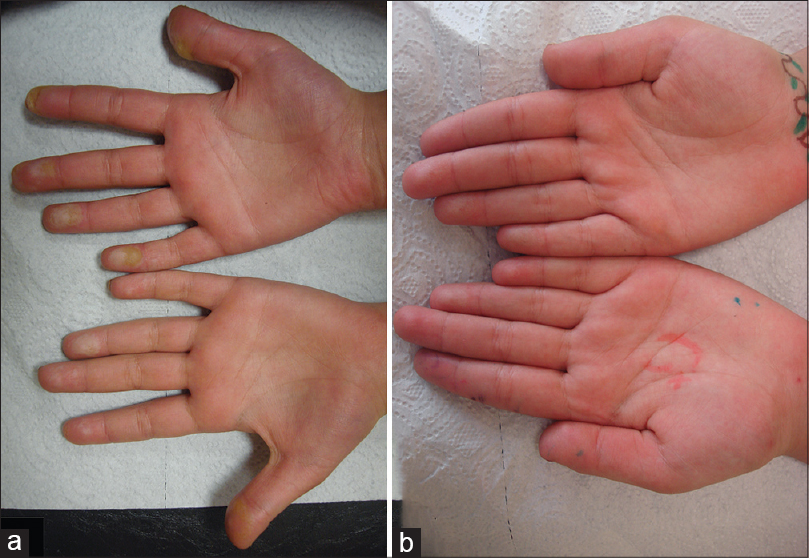 |
| Figure 2: (a) Plaques on the fingertips, (b) showing regression 3 weeks after the patient started a tyrosine-restricted diet |
Oculocutaneous tyrosinemia was suspected, which was supported by her elevated blood and urinary tyrosine levels (blood tyrosine level: 1526 μmol/L, normal: ≤100 μmol/L; urinary tyrosine level: 521 mmol/mol creatinine, normal: ≤58 mmol/mol creatinine) measured at baseline (the patient′s parents and sibling had normal blood and urinary tyrosine levels). Sequencing of the complete coding region of the cytoplasmic tyrosine aminotransferase (TAT) gene revealed the known mutation c.355C>T (p.R119W) (the mutation was homozygous in the patient and heterozygous in both parents). This confirmed the diagnosis of oculocutaneous tyrosinemia. A modified diet with strict restriction of tyrosine and phenylalanine was started. The patient reported significant improvement of her skin lesions and pain within the first week of treatment. At the 3-week follow-up, almost complete regression of the hyperkeratotic plaques was noted [Figure - 1]b and [Figure - 2]b. During the 1.5 years after diagnosis, the patient has largely adhered to the recommended diet and remained symptom-free, except for brief periods of non-adherence followed by mild exacerbations.
Oculocutaneous tyrosinemia is a rare genetic disorder of tyrosine metabolism with autosomal recessive transmission. Among the tyrosinemias, oculocutaneous tyrosinemia is the only form with typical hyperkeratotic lesions of the palmoplantar area. They usually arise as bullae and erosions during childhood and progress to sharply demarcated, yellowish plaques limited to pressure points. Intense pain is an important diagnostic clue. Patients usually present with ocular findings such as photophobia and corneal ulcers in the first year of life. Mental retardation is a highly variable feature. [1] Our patient had normal intelligence (intelligence quotient = 110, Cattell Culture Fair Intelligence Test).
Various dermatological, ophthalmological, and neurological findings of oculocutaneous tyrosinemia have been reported. Lesions of the skin and eyes are probably caused by deposition of tyrosine crystals leading to an inflammatory response. [3] The disorder can be diagnosed by the detection of a high plasma tyrosine level and a normal plasma phenylalanine level, and can be confirmed by demonstrating a mutation in the tyrosine aminotransferase gene located on chromosome 16q22.1-q22.3. [4] Dietary restriction of tyrosine and phenylalanine is a highly effective treatment. Our patient′s daily protein consumption was modified with 0.6 g/kg/day of natural proteins and 0.6 g/kg/day of phenylalanine and tyrosine-restricted special formula. She was advised to abstain from excessive amounts of protein-rich foods such as meat, milk, and egg products. Following these dietary principles, her symptoms were under control while maintaining an adequate and balanced intake of nutrients for growth.
Timely diagnosis can help in the regression of lesions, reduce pain, and save vision, and thus greatly improve the patient′s quality of life. In addition, mental retardation can gradually progress if the patient is not treated. [5] The coexistence of painful keratoderma with ocular symptoms should lead physicians to suspect a diagnosis of oculocutaneous tyrosinemia.
ACKNOWLEDGMENT
The authors thank the patient and her family for their participation in the study. RCB is recipient of a Heisenberg Professorship from the German Research Foundation (DFG).
| 1. |
Krol AL, Siegel D. Keratodermas. In: Bolognia JL, Jorizzo JL, Schaffer JV, editors. Dermatology. 3 rd ed. China: Elsevier; 2012. p. 871-85.
[Google Scholar]
|
| 2. |
Gokhale NS, Dherai AJ, Desai H, Ashavaid TF. Unusual dendritic keratitis. Indian J Ophthalmol 2007;55:57-9.
[Google Scholar]
|
| 3. |
Valikhani M, Akhyani M, Jafari AK, Barzegari M, Toosi S. Oculocutaneous tyrosinaemia or tyrosinaemia type 2: A case report. J Eur Acad Dermatol Venereol 2006;20:591-4.
[Google Scholar]
|
| 4. |
Madan V, Gupta U. Tyrosinaemia type II with diffuse plantar keratoderma and self-mutilation. Clin Exp Dermatol 2006;31:54-6.
[Google Scholar]
|
| 5. |
Scott CR. The genetic tyrosinemias. Am J Med Genet C Semin Med Genet 2006;142:121-6.
[Google Scholar]
|
Fulltext Views
5,906
PDF downloads
1,754





![[Figure - 1]](#fig_ijdvl_2015_81_3_303_152744_f1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2015_81_3_303_152744_f2.jpg){kind=link}