Translate this page into:
Phakomatosis pigmentovascularis Type IIb, Sturge– Weber syndrome and cone shaped tongue: An unusual association
2 Department of Paediatrics, Veer Surendra Sai Medical College, Sambalpur, Odisha, India
Correspondence Address:
Swetalina Pradhan
Qr. No. D/6, Near PWD Offi ce, Burla, Sambalpur - 768 017, Odisha
India
| How to cite this article: Pradhan S, Patnaik S, Padhi T, Nayak BP. Phakomatosis pigmentovascularis Type IIb, Sturge– Weber syndrome and cone shaped tongue: An unusual association. Indian J Dermatol Venereol Leprol 2015;81:614-616 |
Sir,
Phakomatosis pigmentovascularis is a rare syndrome characterized by coexistence of capillary malformation and pigmentary nevus in the same patient. Type II is the most commonly reported subtype of phakomatosis pigmentovascularis.[1] Sturge–Weber syndrome is a rare, sporadic disease characterized by meningofacial angiomatosis with cerebral calcification. We report a case of phakomatosis pigmentovascularis Type IIb and Sturge–Weber syndrome with some atypical features and an unusual association.
A 13-year-old male child born of nonconsanguineous marriage presented to our dermatology outpatient department with dusky red patches bilaterally over face, hands and chest since 5 years. The patient had also noticed enlargement of both lips since last 2½ years. He gave a history of self-resolving right sided hemiparesis 3 years back. Cutaneous examination revealed bilateral well-defined dusky red patches involving the face (in the distribution of all three divisions of trigeminal nerve) upper extremity, chest and oral mucosa (nevus flammeus) [Figure - 1]a. A hypopigmented patch (nevus anemicus) was present over right side of chest and nevus of ota left side and scleral pigmentation right side [Figure - 1]b, [Figure - 1]c and [Figure - 2]a. There was hypertrophy of the lips and jaw with gum hyperplasia and cone shaped tongue [Figure - 1]a. There was disproportionate enlargement of the right forearm, arm and fingers of the right hand compared to left side [Figure - 2]b. Neurological examination revealed moderate intellectual disability, decreased power (Grade IV medical research council scale) and hypertonia in right upper and lower limb. Ocular examination was normal except for scleral pigmentation. Skin biopsy from the lesion on the forearm showed proliferation and ectasia of thin-walled vessels of varying caliber suggesting capillary malformation. Computed tomography scan and magnetic resonance imaging of the brain showed 'tram-track' like calcification in the left parieto-occipital cortex [Figure - 3]a and [Figure - 3]b. Ultrasonogram and X-ray of the abdomen were normal. Based upon clinical, histopathological and radiological findings, a diagnosis of phakomatosis pigmentovascularis Type IIb and Sturge–Weber syndrome in association with cone shaped tongue was made.
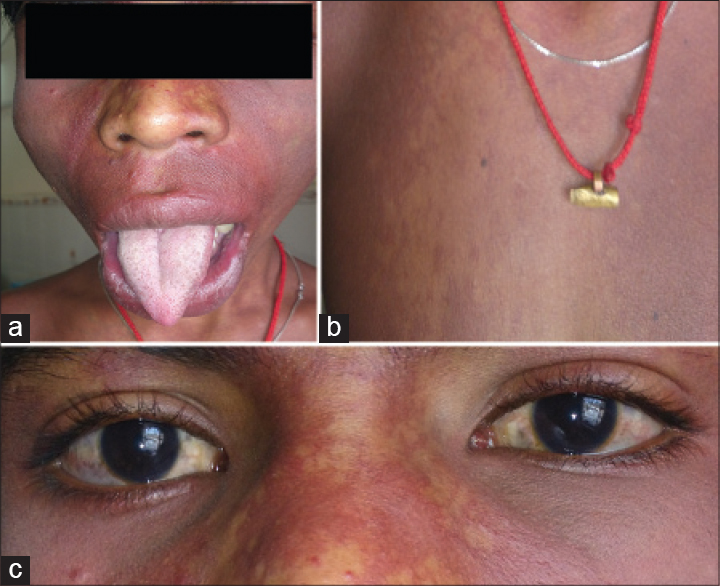 |
| Figure 1: (a) Port-winestain covering bilateral face with hypertrophy of lips, jaw and cone shaped tongue, (b) nevus anemicus, (c) scleral pigmentation |
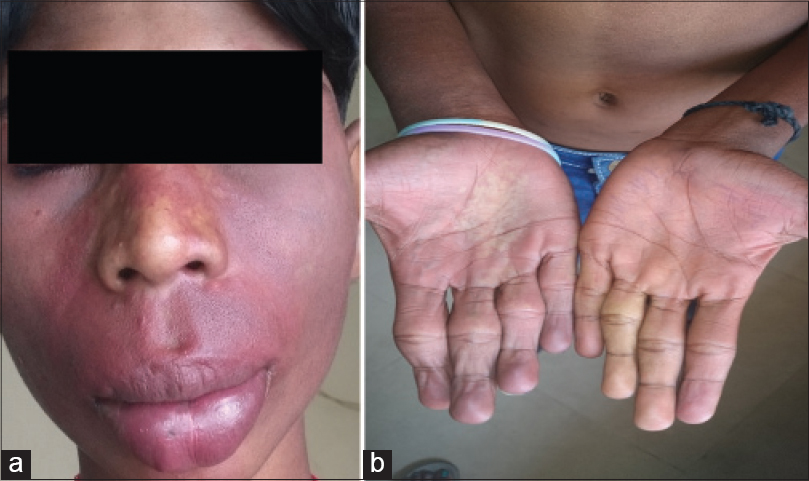 |
| Figure 2: (a) Bluish grey pigmentation over left cheek (b) hypertrophy of fingers of right hand |
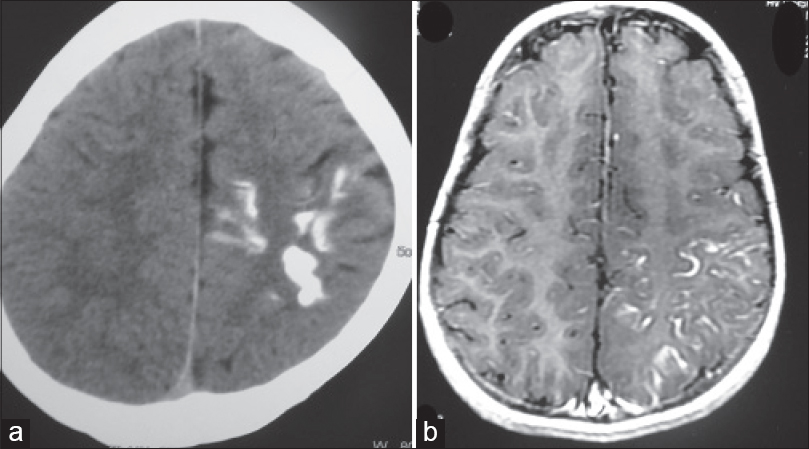 |
| Figure 3: (a) Computed tomography scan of brain showing tram track like calcification over left parietooccipital region, (b) magnetic resonance imaging of brain calcification over left parietooccipital region |
Phakomatosis pigmentovascularis represents a rare cutaneous congenital malformation syndrome characterized by the co-existence of capillary malformation and pigmentary nevi.[1] It was first described by Ota in 1947, as an association of dermal melanocytosis with congenital vascular nevi, mainly capillary malformations. It is sub-classified into 4 types, all having nevus flammeus in common.[2] In addition, Type I has nevus pigmentosus/verrucosus; Type II: Mongolian spots; Type III: nevus spilus and Type IV: Mongolian spots and nevus spilus. Type II–IV may be associated with nevus anemicus.[2] Each type is further subdivided according to the presence of manifestations in skin only (subtype a), or if associated with systemic manifestations (subtype b), which may be in the form of ocular, vascular, skeletal and neurological.[3] Happle in 2005 reclassified phakomatosis pigmentovascularis into three types: phakomatosis cesioflammea (blue spots and nevus flammeus); phakomatosis spilorosea (nevus spilus coexisting with a pale-pink telangiectatic nevus); and phakomatosis cesiomarmorata (blue spots and cutis marmorata telangiectatica congenita).[4] The pathogenesis of phakomatosis pigmentovascularis is still not clear and it has been suggested that it may reflect twin spotting phenomenon (didymosis) due to hypothetic allelic mutation presenting as paired melanocytic and achromic macules or nevus vascularis mixtus.[5]
Sturge–Weber syndrome is a mesodermal phakomatosis characterized by port-wine nevus covering face and cranium in the distribution of the first division of the trigeminal nerve along with atrophy and calcification of the cerebral hemisphere homolateral to the skin lesion and is associated with glaucoma, seizures, paresis and neuro-developmental delay.
Our patient had a history of hemiparesis, port-wine stain (nevus flammeus), nevus of Ota, bluish-gray pigmentation over sclera, nevus anemicus, hypertrophy of gum, lips, right upper limb and fingers with moderate intellectual abnormality, decreased power and hypertonia in the right limbs and tram track calcification in the left parieto-occipital area of brain which were consistent with a diagnosis of phacomatosis pigmentovascularis Type IIb and Sturge–Weber syndrome.
An unusual feature in our patient was that though the port-wine stain covered a large surface area bilaterally, tram track-like calcification was present only on the left side of the cerebral hemisphere. The patient also had an abnormal cone shaped tongue. We were unable to find previous reports of a similar presentation.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
| 1. |
Chang BP, Hsu CH, Chen HC, Hsieh JW. An infant with extensive Mongolian spot, naevus flammeus and cutis marmorata telangiectatica congenita: A unique case of phakomatosis pigmentovascularis. Br J Dermatol 2007;156:1068-71.
[Google Scholar]
|
| 2. |
Hasegawa Y, Yasuhara M. A variant of phakomatosis pigmentovascularis. Skin Res 1979;21:178-86.
[Google Scholar]
|
| 3. |
Fernández-Guarino M, Boixeda P, de Las Heras E, Aboin S, García-Millán C, Olasolo PJ. Phakomatosis pigmentovascularis: Clinical findings in 15 patients and review of the literature. J Am Acad Dermatol 2008;58:88-93.
[Google Scholar]
|
| 4. |
Happle R. Phacomatosis pigmentovascularis revisited and reclassified. Arch Dermatol 2005;141:385-8.
[Google Scholar]
|
| 5. |
de las Heras E, Boixeda JP, Ledo A, Happle R. Paired melanotic and achromic macules in a case of phacomatosis pigmentovascularis: A further example of twin spotting? Am J Med Genet 1997;70:336-7.
[Google Scholar]
|
Fulltext Views
3,306
PDF downloads
1,269





![[Figure - 1]](#fig_ijdvl_2015_81_6_614_168335_f1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2015_81_6_614_168335_f2.jpg){kind=link}
![[Figure - 3]](#fig_ijdvl_2015_81_6_614_168335_f3.jpg){kind=link}