Translate this page into:
Progressive symmetric erythrokeratoderma: Report of a Chinese family
2 Biological Research Center of Huazhong University of Science and Technology, China
Correspondence Address:
Hong-Bo Yan
Department of Dermatology, Wuhan General Hospital of Guangzhou Command, No. 627, Wuluolu Street, Wuhan
China
| How to cite this article: Yan HB, Zhang J, Liang W, Zhang HY, Liu JY. Progressive symmetric erythrokeratoderma: Report of a Chinese family. Indian J Dermatol Venereol Leprol 2011;77:597-600 |
Abstract
A large pedigree of progressive symmetric erythrokeratoderma is reported. The proband was a 22-year-old male with generalized asymptomatic lesions characterized by symmetrical well-demarcated erythematous hyperkeratotic plaques mainly distributed on the extremities. The proband's parents were also affected, and they were first cousins. Thus, a case of familial progressive symmetric erythrokeratoderma is described.Introduction
Progressive symmetric erythrokeratoderma (PSEK), or Gottron′s syndrome, was first described by Darier in 1911, although it is named after Gottron′s article in 1922. [1] It is characterized by stable, well-defined, erythematous, hyperkeratotic plaques distributed mainly on the knees, elbows, buttocks, head and dorsal surfaces of the hands and feet. The trunk is generally spared. The condition is not congenital but has its onset during early childhood. The plaques may progress during childhood, and then are usually stable. In very occasional cases does regression occur. No specific therapy for PSEK is currently available. Here, we report a Chinese family with PSEK. This five-generation family consisted of 45 individuals. There were 16 affected individuals - seven males and nine females.
Case Report
The kindred investigation exhibited the hallmarks of autosomal-dominant inheritance [Figure - 1]. The proband, a 22-year-old male, was the third-born child of first-degree consanguineous parents who were also clinically affected. He had been noted to have scaling erythema on the soles at 4 months of age. Similar lesions gradually developed in the elbow joints, knee joints, metacarpophalangeal and interphalangeal joints symmetrically when he was 14 years old. When he was 17 years old, lesions developed on the dorsum of the hands and forearms. The patient reported that the color of erythema became darker in summer. He is otherwise fit and well.
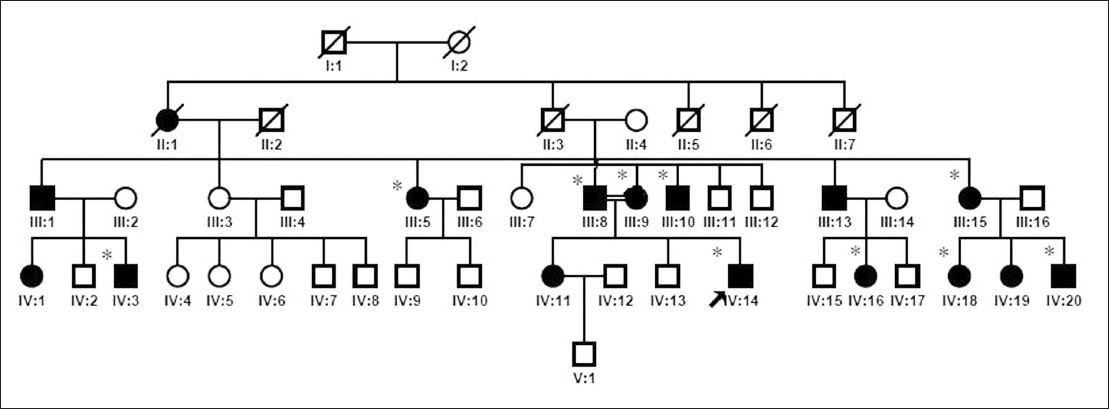 |
| Figure 1: Pedigree tree of progressive symmetric erythrokeratoderma: affected members are represented by black squares; proband is indicated by an arrow (IV-14); individuals signed with asterisk (*) provided sample and were given mycological culture |
Physical examination showed well-demarcated, symmetrically distributed hyperkeratotic erythematous plaques covered with white pityriasiform scales on the extensor surfaces of the extremities, especially on the dorsum of hands and feet, wrists, elbows, knees, ankles and lower limbs close to the ankles. Desquamation and keratosis of the palms and soles were also seen. The nails of the left hand and feet were thickened with a yellowish discoloration, accompanied with transverse ridges in some nail plates. Nails of the right hand were unaffected. His feet were malodorous. Review of the other systems was otherwise normal [Figure - 2].
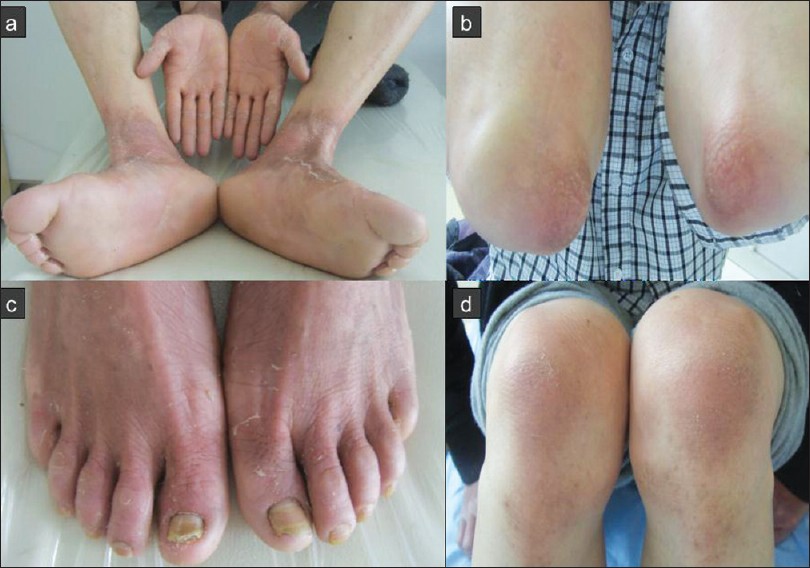 |
| Figure 2: Clinical features: (a, b, d) hyperkeratotic erythematous plaques covered with white pityriasiform scales on the dorsum of the hands and feet, elbows, wrists, knees, ankles and lower limbs close to the ankles. (c) Nail-plate thickening with a yellowish discoloration |
Histological examination of skin biopsy taken from the left forearm showed marked hyperkeratosis, parakeratosis, acanthosis, elongation of rete ridges, extension of dermal papillae and lymphocytic infiltration around the superficial blood vessels [Figure - 3].
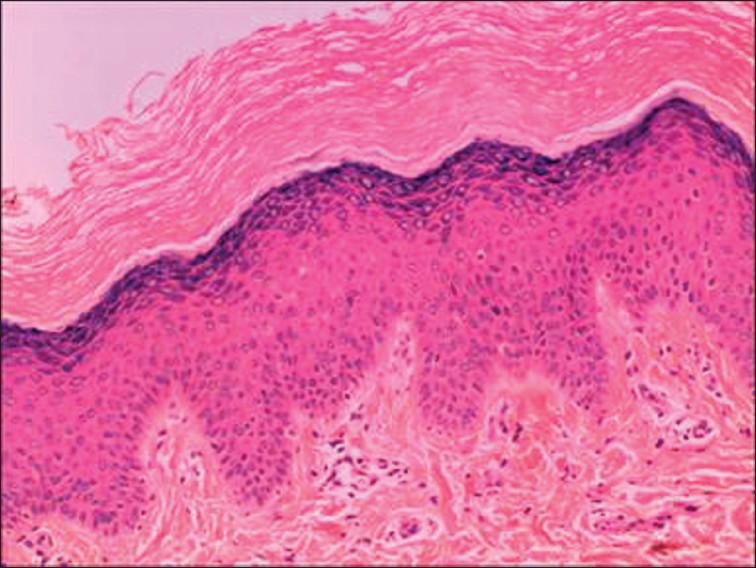 |
| Figure 3: Marked hyperkeratosis and parakeratosis, mild acanthosis, lymphocytic infiltration around the superficial blood vessels (H and E, ×100) |
Based on the clinical and histopathological findings, the proband was diagnosed with PSEK.
The proband showed good response to oral acitretin 20 mg/day along with topical tretinoin 0.025% ointment. A marked improvement with flattening of hyperkeratotic plaques and fading of erythema was observed after 1 month of retinoid therapy. No immediate adverse effect to acitretin was noted. He was doing well on therapy until last follow-up 4 months back.
Discussion
PSEK is a rare genodermatosis with predominantly autosomal-dominant inheritance, although autosomal-recessive transmission has been also observed. It is probable that different clinical presentations may occur within family members, as autosomal-dominant diseases often have incomplete penetrance and variable expressivity. In the family, all patients have typical features of the PSEK trait, but there is significant variable expressivity within the family, especially in coverage of lesions. Most patients were affected only in the hands and feet, whereas the proband was the only one who had generalized lesions. The clinical features of the proband started earlier and the skin lesions were much more pronounced and extended, which might be linked to intermarriage of the proband′s affected parents. Further studies should be conducted with a genetic analysis about this possible association. Furthermore, the proband′s affected sister was reported to have a mild clinical manifestation, probably suggesting underlying genetic heterogeneity.
Because palmoplantar erythema and nail-plate thickening with a yellowish discoloration are common signs of patients in this family, 10 patients with abnormal nails had their nails sent for examination. Samples were collected from the diseased distal nail plate and sent for mycology and culture. Candida albicans and Aspergillus were isolated from the proband′s and his father′s nails, respectively. No fungi were found in the other patients′ culture. Results of our study showed that the prevalence of onychomycosis among PSEK-diseased nails is 20%, which is slightly higher than the prevalence of onychomycosis (16.6%) among the general population in a Chinese study. [2] Although there is no literature reported about mycological prevalence in diseased nails of PSEK, recurrent fungal infections is one of the characteristics of KID (keratitis, ichthyosis, deafness) /HID (hystrix-like, ichthyosis, deafness) syndrome. [3]
Erythrokeratodermas have been divided into two major non-syndromic types [4] and some syndromes such as KID [5],[6] and HID syndrome. The two major non-syndromic types have been described as PSEK and erythrokeratoderma variabilis (EKV). As for PSEK lesions, it is necessary to make a differential diagnosis from other types of erythrokeratodermas, especially EKV. There is considerable similarity to the clinical picture of EKV, with symmetrically distributed, fixed or very slowly progressive erythematous, scaly plaques. It differs in the absence of migratory erythematous lesions and in a greater incidence of palmoplantar keratoderma. Also, as the name would suggest, the symmetry of the lesions in PSEK is more striking than in EKV. In both conditions, the general health is usually not affected. Two sisters have been described by Macfarlane et al., [7] in which the younger one suffered from EKV whereas the older one suffered from PSEK, and this report brought forward the hypothesis that PSEK and EKV were different manifestations of a single condition. Although some cases have been identified to have loricrin and few to have connexin mutations in PSEK, EKV or both, [8],[9],[10] recent reports show that the same gene mutations were not found in some pedigree of PSEK. [11],[12],[13] The other case of a large Chinese pedigree with PSEK was reported in 2004. That seven-generation family consists of 16 affected individuals. The proband was a 16-year-old female. Most patients in that family were affected only in the hands and feet, whereas few patients had generalized lesions spreading to the wrist, forearm, ankle and trunk. The authors also probed the causative role of the loricrin and connexin gene, but excluded the pathogenic mutations in the family, which indicates that PSEK is a heterogeneous disorder and probably has some new genetic locus that is responsible. [13] Thus, accumulating genetic studies support the presumption that there is distinct molecular pathology between these two subtypes of erythrokeratodermas.
In conclusion, a new family of PSEK is reported, with proband′s parents being clinically affected first cousins. The similar condition was reported in a Turkish family, with the proband′s parents also being first cousins but clinically unaffected. [11]
Acknowledgments
This work was funded by grants from the Natural Science Foundation of Hubei province (Grant number: 2010CDB09203).
| 1. |
Gottron HA. Congenital symmetrical progressive erythrokeratoderma. Arch Dermatol Syph 1923;7:416.
[Google Scholar]
|
| 2. |
Cheng S, Chong L. A prospective epidemiological study on tinea pedis and onychomycosis in Hong Kong. Chin Med J (Engl) 2002;115:860-5.
[Google Scholar]
|
| 3. |
Van Steensel MA. Gap junction diseases of the skin. Am J Med Genet C Semin Med Genet 2004;131C:12-9.
[Google Scholar]
|
| 4. |
Bongiorno MR, Aricò M. Progressive symmetric erythrokeratodermia associated with oligodontia, severe caries, disturbed hair growth and ectopic nail: A new syndrome? Dermatology 2008;217:347-50.
[Google Scholar]
|
| 5. |
Richard G, Rouan F, Willoughby CE, Brown N, Chung P, Ryynänen M, et al. Missense mutations in GJB2 encoding connexin-26 cause the ectodermal dysplasia keratitis-ichthyosis-deafness syndrome. Am J Hum Genet 2002;70:1341-8.
[Google Scholar]
|
| 6. |
Van Steensel MA, van Geel M, Nahuys M, Smitt JHS, Steijlen PM. A novel connexin 26 mutation in a patient diagnosed with keratitis-ichthyosis-deafness syndrome. J Invest Dermatol 2002;118:724-7.
[Google Scholar]
|
| 7. |
Macfarlane AW, Chapman SJ, Verbov JL. Is erythrokeratoderma one disorder? A clinical and ultrastructural study of two siblings. Br J Dermatol 1991;124:487-91.
[Google Scholar]
|
| 8. |
Ishida-Yamamoto A, McGrath JA, Lam H, Iizuka H, Friedman RA, Christiano AM. The molecular pathology of autosomal dominant erythrokeratoderma: A frameshift mutation in the loricrin gene and perturbations in the cornified cell envelope. Am J Hum Genet 1997;61:581-9.
[Google Scholar]
|
| 9. |
Ishida-Yamamoto A. Loricrin keratoderma: A novel disease entity characterized by nuclear accumulation of mutant loricrin. J Dermatol Sci 2003;31:3-8.
[Google Scholar]
|
| 10. |
Van Steensel MA, Oranje AP, van der Schroeff JG, Wagner A, van Geel M. The missense mutation G12D in connexin30.3 can cause both erythrokeratodermia variabilis of Mendes da Costa and progressive symmetric erythrokeratodermia of Gottron. Am J Med Genet A 2009;149:657-61.
[Google Scholar]
|
| 11. |
Akman A, Masse M, Mihci E, Richard G, Christiano AM, Balle BJ, et al. Progressive symmetrical erythrokeratoderma: Report of a Turkish family and evaluation for loricrin and connexin gene mutations. Clin Exp Dermatol 2008;33:582-4.
[Google Scholar]
|
| 12. |
Wei S, Zhou Y, Zhang TD, Huang ZM, Zhang XB, Zhu HL, et al. Evidence for the absence of mutations at GJB3, GJB4 and LOR in progressive symmetrical erythrokeratodermia. Clin Exp Dermatol 2011;36:399-405.
[Google Scholar]
|
| 13. |
Cui Y, Yang S, He PP, Zhou WM, Li M, Gao M, et al. Progressive symmetric erythrokeratodermia: Report of a Chinese family and evidence for genetic heterogeneity. J Dermatol Sci 2004;35:233-5.
[Google Scholar]
|
Fulltext Views
5,771
PDF downloads
2,767





![[Figure - 1]](#fig_ijdvl_2011_77_5_597_84070_u1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2011_77_5_597_84070_u2.jpg){kind=link}
![[Figure - 3]](#fig_ijdvl_2011_77_5_597_84070_u3.jpg){kind=link}