
Translate this page into:
Progressive symmetric erythrokeratodermia with spinocerebellar ataxia due to ELOVL4 mutation in a Chinese family
Corresponding author: Dr. Huijun Wang, Department of Dermatology, Peking University First Hospital, Beijing Key Laboratory of Molecular Diagnosis on Dermatoses, National Clinical Research Center for Skin and Immune Diseases, Beijing, China. drhuijunwang@pku.edu.cn
-
Received: ,
Accepted: ,
How to cite this article: Wang Z, Lin Z, Wang H. Progressive symmetric erythrokeratodermia with spinocerebellar ataxia due to ELOVL4 mutation in a Chinese family. Indian J Dermatol Venereol Leprol 2022;88:132.
Sir,
Erythrokeratodermia refers to a group of inherited skin disorders characterized by coexistence of figurate erythema and hyperkeratotic plaques. It can be subdivided into two main forms, that is, erythrokeratodermia variabilis and progressive symmetric erythrokeratodermia, although a new designation of erythrokeratodermia variabilis et progressiva was proposed due to overlap of the two forms. Erythrokeratodermia with ataxia, or spinocerebellar ataxia 34 (SCA34; OMIM 133190), is an autosomal dominant genetic disease characterized by a combination of erythrokeratodermia and spinocerebellar ataxia. Recently, mutations in ELOVL4 were identified to cause SCA34.1 All the reported SCA34 cases with skin lesions showed phenotype of erythrokeratodermia variabilis or reminiscent of erythrokeratodermia variabilis which is characterized by transient or migratory erythematous plaques.1-4 Herein, we report a Chinese familial case of SCA34 due to ELOVL4 mutation. Interestingly, the patients had slowly progressive erythrokeratotic plaques which were consistent with progressive symmetric erythrokeratodermia rather than erythrokeratodermia variabilis.
A 29-year-old man of Chinese Han ethnicity presented with reddish raised lesions on limbs. First noted at the age three years, the skin lesions progressed gradually during childhood and stabilized after adolescence. Pathognomonic symptoms of erythrokeratodermia variabilis, such as transient reddish lesions, were denied. He developed mild weakness of lower limbs and walking disability one year ago. Physical examination revealed symmetrically distributed and sharply demarcated erythematous hyperkeratotic plaques on his wrists, elbows and lower limbs [Figure 1]. Histopathology of the affected skin showed non-specific findings with orthokeratosis, acanthosis and superficial dermal infiltration of monocytes. No abnormality was found in the craniocerebral computed tomography scan. The proband’s 54-year-old mother had similar but milder skin lesions since childhood which were persisting. She developed slowly progressive gait ataxia in the fourth decade. Her brain magnetic resonance imaging showed moderate cerebellar and pontine atrophy.
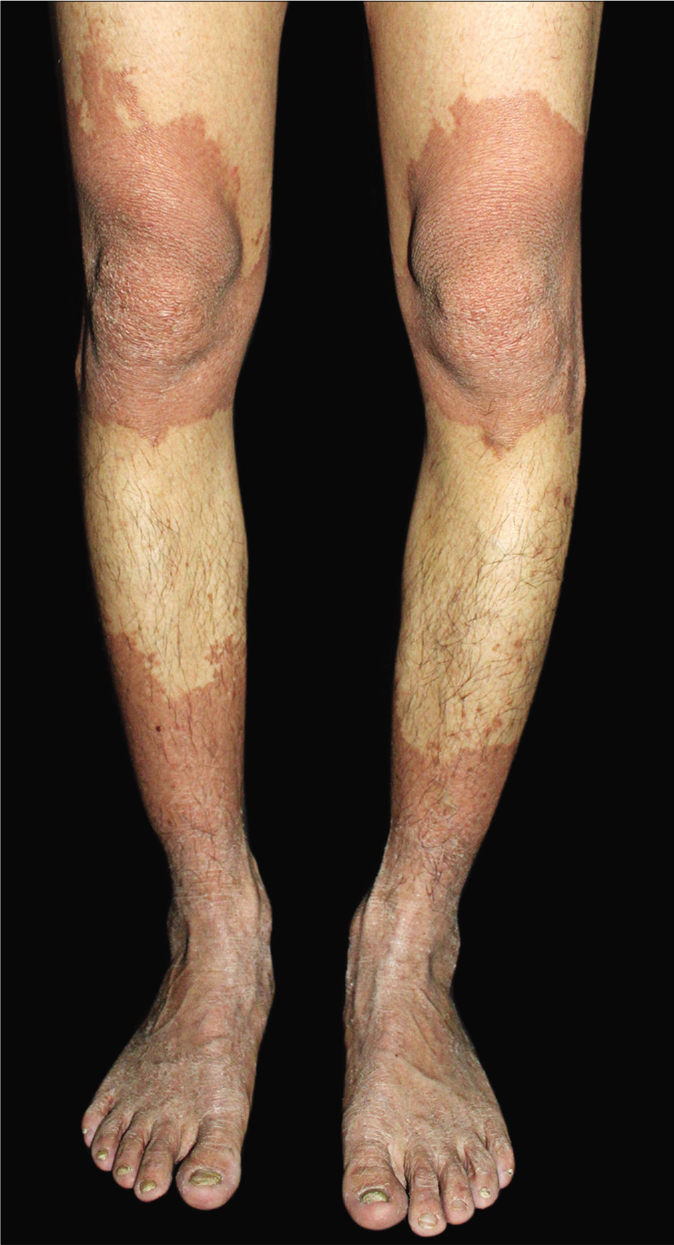
- The proband with symmetrically distributed and sharply demarcated erythematous hyperkeratotic plaques on knees, calves, ankles, feet and elbows, sparing the trunk
To determine the underlying genetic cause, blood samples of the proband, his parents and grandmother were collected after obtaining written informed consent. Whole-exome sequencing was performed in the proband which revealed a heterozygous mutation of c.698C>T (p.T233M) in the ELOVL4 gene. Sanger sequencing verified this mutation in the proband and his mother [Figures 2a and 2b]. The affected residue threonine 233 located within the sixth transmembrane helix of ELOVL4 protein is a highly conserved amino acid. This mutation was predicted to be “damaged” by Polyphen-2 and Mutationtaster.3
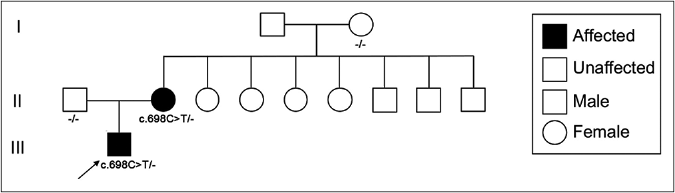
- Pedigree of the SCA34 kindred with mutation (c.698C>T) in ELOVL4. Arrow indicates the proband in the family
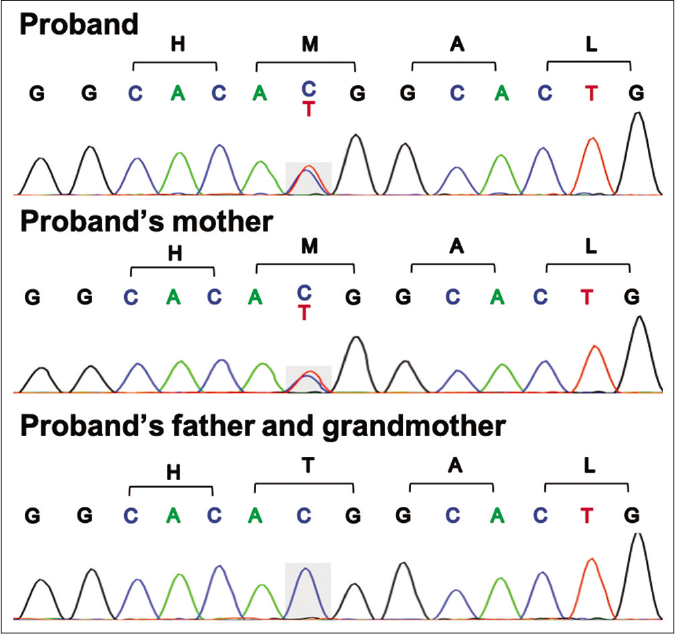
- Sanger sequencing of ELOVL4 revealed heterozygous missense mutation c.698C>T (p.T233M) in the proband and his mother which was not found in his father and grandmother
We herein report the first Chinese case of SCA34 caused by mutations in ELOVL4. Expressed in brain, skin, retina and testes, ELOVL4 encodes a specific fatty acid elongase which catalyzes the rate-limiting reaction in the elongation of very long-chain fatty acids.1 These fatty acids are subsequently utilized for the biosynthesis of lipid substances including ceramides, a well-known necessary component for skin barrier formation.
To date, only 4 ELOVL4 mutations have been reported in six cases of SCA34 [Figure 2c].1-5 Although not all cases of ELOVL4-associated SCA34 presented with skin lesions,3 all reported SCA34 patients with skin lesions manifested as erythrokeratodermia variabilis or localized erythrokeratodermia.1,2 Interestingly, the proband in this study had slowly progressive erythrokeratotic plaques which became stationary after adolescence. His skin lesions were consistent with progressive symmetric erythrokeratodermia. We were unable to find any previous reports of this condition in SCA34 patients, although we could not exclude the possibility that similar skin conditions were described as erythrokeratodermia variabilis unwittingly since all the previous cases were published in neurological journals.
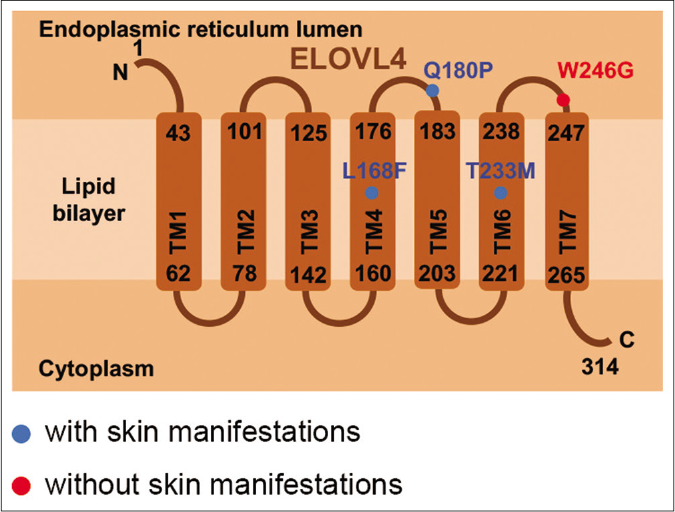
- Structure schematic of ELOVL4. All the reported mutational sites leading to SCA34 are shown. TM: Transmembrane domain, N: Aminoterminus, C: Carboxyterminus
The mutation c.698C>T in ELOVL4 is a recurrent mutation that has previously been reported in two cases.3,4 One was a 60-year-old English-Canadian female who had widespread variable itchy erythematous keratotic plaques since the age of four years.4 The other case was a Japanese kindred, the proband showed late-onset erythematous keratotic plaques on bilateral ankles since the age of 55 years, while his father, harboring the same mutation, had no skin lesion.3 The reason why the same mutation is linked to various skin manifestations remains elusive and was speculated to be relative to race, nursing conditions or environmental factors.
The onset of ataxia in SCA34 mostly occurs in the fourth or fifth decade of life,1 so it’s reasonable that the proband here exhibited only subtle neurological manifestations, while his affected mother has shown organic brain impairment. Nevertheless, the proband is very likely to become symptomatic eventually, so long-term follow-up by a neurologist is necessary. This also reminds us that when a young patient presents with erythrokeratodermia, SCA34 should also be considered as a differential diagnosis, even when neurological symptoms are absent.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Financial support and sponsorship
This study was supported financially by Peking University Medicine Fund of Fostering Young Scholar’s Scientific and Technological Innovation (grant no. BMU2020PY006).
Conflicts of interest
There are no conflicts of interest.
References
- Expanding the clinical phenotype associated with ELOVL4 mutation: Study of a large French-Canadian family with autosomal dominant spinocerebellar ataxia and erythrokeratodermia. JAMA Neurol. 2014;71:470-5.
- [CrossRef] [PubMed] [Google Scholar]
- A new ELOVL4 mutation in a case of spinocerebellar ataxia with erythrokeratodermia. JAMA Neurol. 2015;72:942-3.
- [CrossRef] [PubMed] [Google Scholar]
- Prevalence and clinicoradiological features of spinocerebellar ataxia Type 34 in a Japanese ataxia cohort. Parkinsonism Relat Disord. 2019;65:238-42.
- [CrossRef] [PubMed] [Google Scholar]
- Novel ELOVL4 mutation associated with erythrokeratodermia and spinocerebellar ataxia (SCA 34) Neurol Genet. 2018;4:e263.
- [CrossRef] [PubMed] [Google Scholar]
- A novel mutation in ELOVL4 leading to spinocerebellar ataxia (SCA) with the hot cross bun sign but lacking erythrokeratodermia: A broadened spectrum of SCA34. JAMA Neurol. 2015;72:797-805.
- [CrossRef] [PubMed] [Google Scholar]




