Translate this page into:
Silvery hair with bronze-tan in a child: A case of Elejalde disease
Correspondence Address:
Arun C Inamadar
Department of Dermatology, Venereology and Leprosy, BLDEA's SBMP Medical College, Hospital and Research Centre, Bijapur - 586 103, Karnataka
India
| How to cite this article: Inamadar AC, Palit A. Silvery hair with bronze-tan in a child: A case of Elejalde disease. Indian J Dermatol Venereol Leprol 2007;73:417-419 |
Abstract
A 5-year-old boy was admitted for severe neurological impairment including hypotonia and loss of consciousness without preceding febrile illness. On examination, he had silver colored hair and bronze-tan over photo-exposed body parts. He was born of consanguineous parents and three of his elder siblings, who died in early childhood, had similar colored hair. Complete blood count and serum immunoglobulin levels were within normal limits. Peripheral blood smear did not show any cytoplasmic granules in neutrophils. Cerebro-spinal fluid examination did not reveal any abnormality. Light microscopic examination of the hair revealed irregular clumping of the melanin throughout the shafts. The patient died on the second day following admission. A clinical diagnosis of Elejalde disease was made. The clinical and genetic overlapping of the three silvery-hair syndromes has been discussed.
 |
| Figure 2: Patient�s hair showing characteristic irregular clumps of melanin (x200) |
|
| Figure 2: Patient�s hair showing characteristic irregular clumps of melanin (x200) |
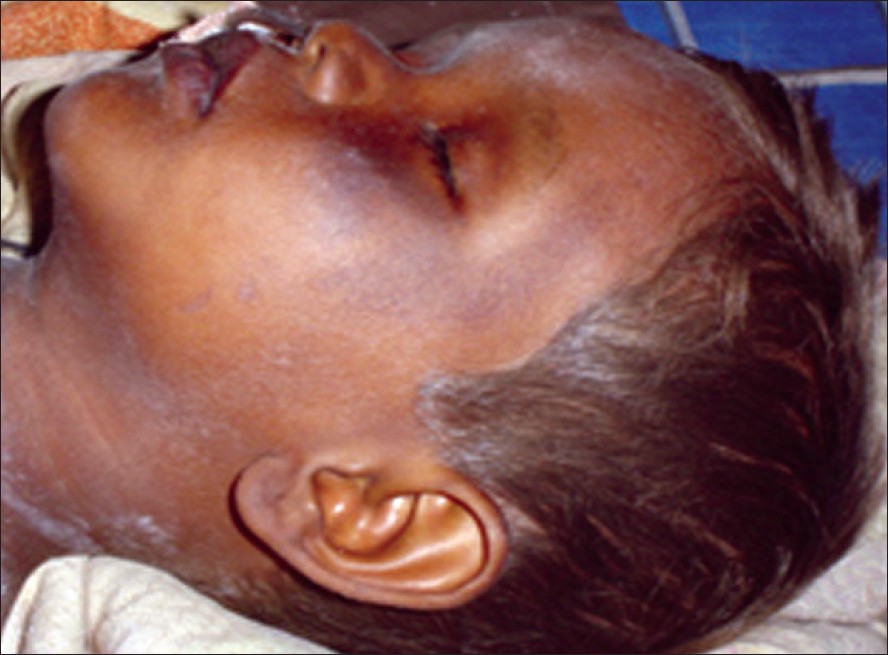 |
| Figure 1: Silvery hair with bronze-tan over face in Elejalde disease |
|
| Figure 1: Silvery hair with bronze-tan over face in Elejalde disease |
Introduction
Elejalde disease (ED) (OMIM entry no. 256710) is a rare neuroectodermal melanolysosomal disorder with autosomal recessive inheritance. [1] Pigmentary abnormalities, skin tanning on sun-exposure and profound disturbances of the central nervous system are characteristic of this disorder. It constitutes a part of the ′silvery hair syndrome′ in association with two other conditions, Chediak-Higashi syndrome (CHS) and Griscelli syndrome (GS). Majority of the reported cases of ED are from Mexico. [1],[2] Here, an Indian boy with a clinical diagnosis of ED has been reported.
Case Report
Dermatological consultation was asked for a 5-year-old boy, admitted under pediatric neurology unit for altered consciousness and generalized hypotonia of 2 weeks′ duration. His parents had noticed a gradual alteration in his complexion and hair color for the past 2 years. Since the past 2 weeks, he had developed a gradual weakness of all the four limbs followed by altered sensorium. There was no preceding febrile illness or convulsion. With progressive loss of consciousness, he was brought to the hospital. He did not have any major/ recurrent infections or other illnesses in the past.
On clinical examination, the child was found to be deeply comatose with loss of reflexes in all the four limbs. Cutaneous examination revealed slightly scaly, bronze-colored patches over the butterfly area of face and distal extremities [Figure - 1]. Skin color of the covered body parts was similar to that of other family members. Eyebrows and the scalp hair along the frontal hair-line showed a silvery shine [Figure - 1].
The patient was born of consanguineous parents and he had six elder siblings. He was apparently normal at birth and thereafter, till the present illness started. His parents, the only alive elder brother or other family members did not have any cutaneous or neurological disorder. Five of the siblings had died by the 4 th to the 6 th year of age. Out of these five siblings, two boys and the only girl had silver-tinged hair. The exact causes of death of these children could not be elicited.
Ophthalmoscopic examination did not reveal any abnormality. Complete blood count was normal. Peripheral blood smear did not show any cytoplasmic granules in neutrophils. Serum immunoglobulin levels and the biochemical parameters were within normal limits. Cytological and biochemical examination of the cerebro-spinal fluid and culture for organisms did not reveal any abnormality. A computerized tomographic scan of the brain revealed diffuse cerebral edema. A few silver-colored hairs were plucked and examined under light microscope. It revealed irregular clumping of the melanin throughout the hair-shafts [Figure - 2].
A combination of features like silver-colored hair, neurological involvement, irregular clumps of melanin in the hair-shaft, in the absence of immunological abnormalities and cytoplasmic granules in the neutrophils led to the diagnosis of ED. The patient was managed with basic life care support and treatment was given to reduce cerebral edema. There was no significant clinical improvement. A skin biopsy was planned for light microscopic and ultrastructural studies, but the patient′s condition deteriorated further. He died on the second day of admission due to sudden cardio-respiratory arrest.
Discussion
Elejalde disease is a rare disorder, with less than 20 cases reported in the US. [1] It was first described by Elejalde et al , in the late 1970s. [3] Neurological manifestations are the predominant clinical features in these patients, which usually appear early, at birth or during childhood. Seizures, severe hypotonia and mental retardation are the usual clinical findings. [4] In advanced disease, flaccid or spastic hemiplegia, quadriplegia and ataxia have been reported. Ophthalmologic abnormalities include nystagmus, diplopia, amaurosis and absence of pupillary reflex. [4] Pigmentary abnormalities are the second common clinical feature which include silvery hair, a gradual bronze-colored tan of the sun-exposed areas and generalized hypopigmentation of covered body parts. Early death due to neurological involvement is a rule and the oldest patient reported with ED was 12 years old. [5]
Elejalde disease is distinct from two other conditions with silvery hair, CHS and GS. Cell-mediated and humoral immune response abnormalities are observed in GS and CHS, along with impaired neutrophil chemotaxis in the latter, which are not seen in ED. Hence, recurrent infective episodes are common in the former two conditions. Oculocutaneous pigment dilution is a prominent feature in CHS; generalized cutaneous hypopigmentation may sometimes be seen in GS and ED. An accelerated phase of illness with lymphoma-like infiltration of different organ systems leading to death occurs in some patients with CHS, [2] and also in GS. [4] This feature is not observed in ED. Certain laboratory investigations can also differentiate these three entities [Table - 1].
However, there is some degree of clinical and genetic overlap between GS, CHS and ED. All three disorders manifest during childhood and central nervous system involvement is common. Inheritance pattern is autosomal recessive for these disorders. Genetically, there is a similarity between GS and ED and some authors have considered ED as an allelic variant of GS. [6] Impaired melanosome transport, giving rise to failure of transfer of melanin to keratinocytes, results in the pigmentary abnormalities in patients with silvery hair syndrome, [7] and the affected genes are believed to be involved in lysosomal transport of melanosomes. [2] The two closely related genes for GS are located on chromosome 15q21. [2],[8] These genes, RAB27A and MYO5A, function in vesicle trafficking at the cellular level. [8] Mutations in RAB27A result in hemophagocytic abnormalities and thereby result in GS type 2, manifested as silvery hair with immunological defects. Mutations in MYO5A (an actin-based motor molecule, required for pigmentation and synaptic activity in the central nervous system) [9] result in pigmentary dilution along with neurological abnormalities, designated as GS type 1. The genetic defect in ED has not been confirmed yet, but some authors believe that GS1, clinically as well as genetically, is equivalent to ED. [8],[10],[11],[12]
Though immunological abnormalities are not seen in ED, in the series of patients reported by Duran-McKinster et al , [2] one had persistent leucopenia and the authors presumed that a subset of ED with immune compromise may exist. A patient of ED who died of respiratory infection has also been reported in the literature. [1] An accelerated phase may also exist regarding the development of neurological manifestations in patients with ED, as observed in our patient and the second, third and sixth patients reported by Duran-McKinster et al . [2] Like our patient, these three children developed normally without any evidence of neurological impairment till a certain age. Thereafter, they had succumbed to fatal, irreversible neurological complications.
A lighter skin color of the covered body parts, contrasting the bronze-tan over photo-exposed parts, was the finding in all patients reported by Duran-McKinster et al . [2] Our patient showed bronze-colored skin over face and distal extremities but a normal skin color like in other family members over the covered body parts. Similarly, the 12-year-old patient with ED, reported by Ivanovich et al , [5] did not show generalized skin color abnormalities except a bronze-tan and diffuse freckling over exposed body parts. Hence, pigmentation abnormalities may be variable among patients with ED.
The present case was a boy of Indian origin who had clinical features of ED and presence of irregular clumping of melanin in hair evidenced by light-microscopy. Absence of hypopigmentation of the covered body parts and an accelerated neurological involvement leading to sudden death in the patient are interesting features.
| 1. |
Scheinfeld NS. Elejalde syndrome. e-Medicine. Available from: http://www.emedicine.com/derm/topic928htm. [Last accessed on 2006 Aug 31].
[Google Scholar]
|
| 2. |
Duran-McKinster C, Rodriguez-Jurado R, Ridaura C, de la Luz Orozco-Covarrubias MA, Tamayo L, Ruiz-Maldonando R. Elejalde syndrome: A melanolysosomal neurocutaneous syndrome: Clinical and morphological findings in 7 patients. Arch Dermatol 1999;135:182-6.
[Google Scholar]
|
| 3. |
Elejalde BR, Holguin J, Valencia A, Gilbert EF, Molina J, Marin G, et al . Mutations affecting pigmentation in man: I. Neuroectodermal melanolysosomal disease. Am J Med Genet 1979;3:65-80.
[Google Scholar]
|
| 4. |
Cahali JB, Fernandez SA, Oliveira ZN, Machado MC, Valente NS, Sotto MN. Elejalde syndrome: Report of a case and review of the literature. Pediatr Dermatol 2004;21:479-82.
[Google Scholar]
|
| 5. |
Ivanovich J, Mallory S, Storer T, Ciske D, Hing A. 12-year-old male with Elejalde syndrome (neuroectodermal melanolysosomal disease). Am J Med Genet 2001;98:313-6.
[Google Scholar]
|
| 6. |
Sanal O, Yel L, Kucukali T, Gilbert-Barnes E, Tardieu M, Texcam I, et al . An allelic variant of Griscelli disease: Presentation with severe hypotonia, mental motor retardation and hypopigmentation consistent with Elejalde syndrome (neuroectodermal melanolysosomal disorder). J Neurol 2000;247:570-2.
[Google Scholar]
|
| 7. |
Lambert J, Vancoillie G, Naeyaert JM. Elejalde syndrome revisited. Arch Dermatol 2000;136:120-1.
[Google Scholar]
|
| 8. |
Anikster Y, Huizing M, Anderson PD, Fitzpatrick DL, Klar A, Gross-Kieselstein E, et al . Evidence that Griscelli syndrome with neurological involvement is caused by mutation in RAB27A, not MYO5A. Am J Hum Genet 2002;71:407-14.
[Google Scholar]
|
| 9. |
Libby RT, Lillo C, Kitamoto J, Williams DS, Steel KP. Myosin Va is required for normal photoreceptor synaptic activity. J Cell Sci 2004;117:4509-15.
[Google Scholar]
|
| 10. |
Menasche G, Fischer A, de Saint Basile G. Griscelli syndrome types 1 and 2. Am J Hum Genet 2002;71:1237-8.
[Google Scholar]
|
| 11. |
Kraft P, Wilson M. Family-based association tests incorporating parental genotypes. Am J Hum Genet 2002;71:1238-9.
[Google Scholar]
|
| 12. |
Bahadoran P, Ortonne JP, Ballotti R, de Saint-Basile G. Comment on Elejalde syndrome and relationship with Griscelli syndrome. Am J Med Genet A 2003;116:408-9.
[Google Scholar]
|
Fulltext Views
4,240
PDF downloads
2,325





![[Figure - 1]](#fig_ijdvl_2007_73_6_417_37063_1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2007_73_6_417_37063_2.jpg){kind=link}
![[Table - 1]](#tbl_ijdvl_2007_73_6_417_37063_3.jpg){kind=link}