Translate this page into:
Sturge-Weber syndrome in association with Klippel-Trenaunay syndrome and phakomatosis pigmentovascularis type IIb
2 R G Kar Medical College, Kolkata, West Bengal, India
3 Department of Radiology, North Bengal Medical College, Darjeeling, West Bengal, India
Correspondence Address:
Rajesh Kumar Mandal
Department of Dermatology, Venereology, and Leprosy, North Bengal Medical College, Darjeeling - 734 012, West Bengal
India
| How to cite this article: Mandal RK, Ghosh SK, Koley S, Roy AC. Sturge-Weber syndrome in association with Klippel-Trenaunay syndrome and phakomatosis pigmentovascularis type IIb. Indian J Dermatol Venereol Leprol 2014;80:51-53 |
Abstract
Phakomatosis pigmentovascularis (PPV) is a rare combination of pigmentary and vascular components with or without systemic involvement. We report here a rare association of Sturge-Weber syndrome, Klippel-Trenaunay syndrome, and PPV type IIb in a 15-year-old boy who had right upper limb monoparesis along with a history of recurrent convulsions.Introduction
Phakomatosis pigmentovascularis (PPV) is a rare association of congenital pigmentary and vascular abnormalities with or without systemic involvement. [1] The pigmentary components comprise dermal melanocytosis including ocular melanosis, aberrant Mongolian spots, nevus of Ota, nevus spilus, and verrucous pigmented nevus. [1] Capillary malformation is the main vascular component of this condition. [2] Various attempts have been made to classify these rare syndromes under the umbrella term, ′phakomatosis pigmentovascularis′. We report here a case of PPV having overlapping features of Sturge-Weber syndrome and Klippel-Trenaunay syndrome for its extreme rarity and a few unusual and interesting manifestations.
Case Report
A 15-year-old boy, born to nonconsanguineous parents, presented with widespread red patches over his face, upper parts of the trunk, and left upper limb. The lesions were present since birth. He was noticed to have gradual enlargement of the right half of his face and the left upper limb for the 5 years prior to consultation. He had a history of recurrent convulsions since his childhood and for the last 3 months he developed progressive weakness of the right upper limb. There was no significant family history. Cutaneous examination revealed a well-defined large erythematous non-blanchable patch over his face, extending over to the neck, chest, upper back, and left upper limb.
There was hypertrophy of the right side of the face with enlarged lower lip and jaw. The left forearm and hand were also disproportionately enlarged in comparison to the opposite side [Figure - 1].
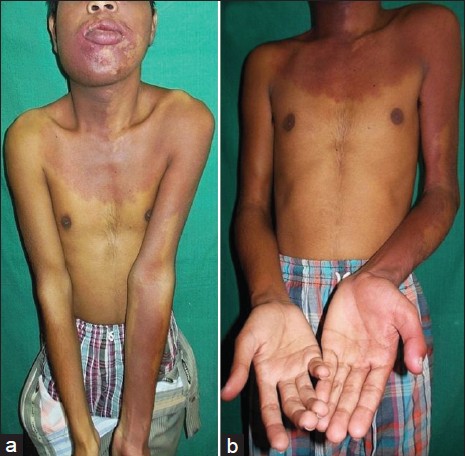 |
| Figure 1: (a) Port-wine stain involving face, neck, chest, and left upper limb. (b) Hypertrophy of left forearm and hand |
In addition, the patient had gum hypertrophy. Oral mucosa was otherwise normal. Blue-colored scleral pigmentation was noted in both eyes, more on the left side [Figure - 2]. Ocular examination was otherwise normal including normal intraocular tension. No lesional bruit or murmur was heard. Neurological assessment showed decreased power (grade III Medical Research Council scale), [3] exaggerated jerks, and increased muscle tone of the right upper limb. Higher functions were normal. There was no sign of cranial nerve involvement or any sensory changes. Bladder and bowel and other autonomic functions were normal. Left hand showed soft tissue and bony hypertrophy on X-ray [Figure - 3]. Ultrasonography (USG) showed hypertrophy of the forearm muscles of the left side in comparison to that of the right. Color Doppler study of upper limbs revealed mild venous varicosities of the left side. X-ray of the skull showed intracranial calcification in the form of a ′tram-track′ and a computed tomography (CT) scan revealed gross atrophy with gyral calcification of left temporoparietal lobe of brain along with cortical thickening [Figure - 4]. Interictal electroencephalography (EEG) was normal. Chest X-Ray and USG of the whole abdomen were normal.
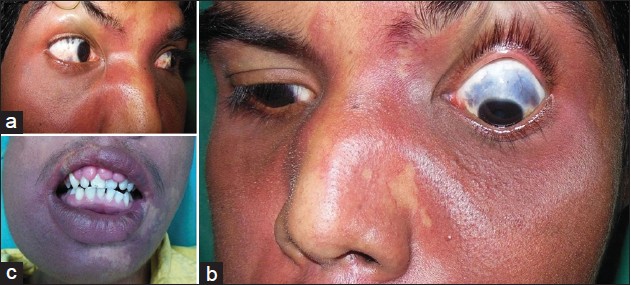 |
| Figure 2: (a and b) Ocular melanosis, (c) Gum hypertrophy |
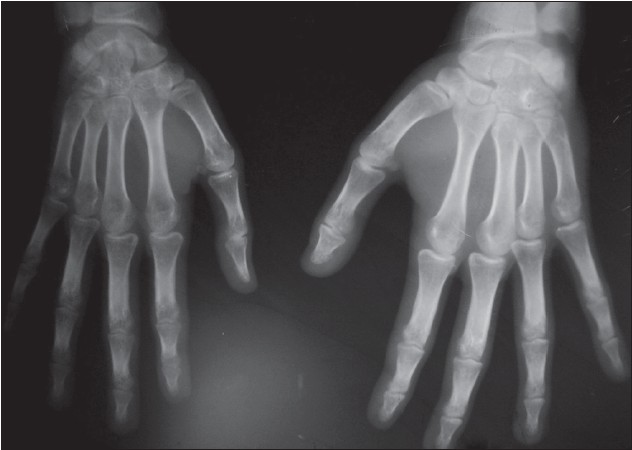 |
| Figure 3: X-ray both hands showing of the soft tissue and bony hypertrophy |
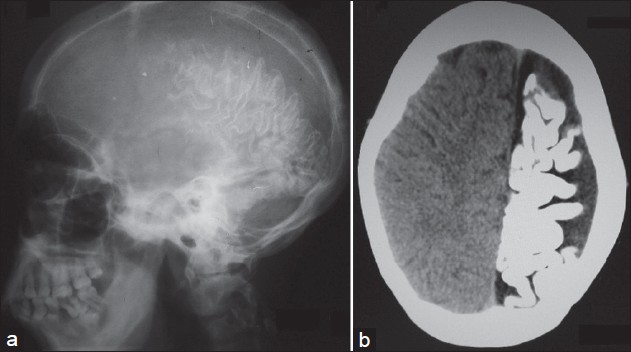 |
| Figure 4: (a) X-ray lateral view of skull showing 'tram-track' calcification. (b) Computed tomography (CT) scan of brain showing atrophy and extensive gyral pattern of calcifi cation of left cerebral hemisphere |
Based on the clinical and radiological features, a diagnosis of PPV type IIb (phakomatosis cesioflammea) associated with Sturge-Weber syndrome and Klippel-Trenaunay syndrome was made.
Dicussion
Phakomatosis pigmentovascularis is a rare cutaneous malformation characterized predominantly by vascular and melanocytic components. [4] PPV has been classified into four subtypes. [2] Each of them either have only cutaneous features (type a) or both cutaneous and extracutaneous features (type b). Port-wine stain (PWS) is present in all the types. Melanocytic components may comprise of dermal melanocytosis including ocular melanosis, aberrant Mongolian spots, nevus of Ota, nevus spilus, and verrucous pigmented nevus. The extracutaneous involvement predominantly causes ocular, skeletal, and neurological alterations. [4],[5],[6],[7],[8],[9] Most of the reported cases of PPV were of type II variety. Vidaurri de-la Cruz et al., reported 24 cases of PPV with an average follow-up period of 5 years out of which 18 were of type IIb and six were of type IIa PPV. [10] In 1987, Ruiz-Maldonado et al., described four cases of PPV and all of them belonged to the type II variety. [11] Types I, III, and IV are rare and only a few cases have been described in the literature.
As the traditional classification was complicated, Happle proposed a more simplified classification of PPV which included three types: phakomatosis cesioflammea (blue spots and nevus flammeus); phakomatosis spilorosea (nevus spilus coexisting with a pale-pink telangiectatic nevus); and phakomatosis cesiomarmorata (blue spots and cutis marmorata telangiectatica congenita). [12] According to this classification, phakomatosis cesioflammea represents the traditional types IIa and IIb; phakomatosis spilorosea is identical to types IIIa and IIIb; and phakomatosis cesiomarmorata describes type V. There is no existence of traditional type I and the traditional type IV is now included in the group of ′unclassifiable′ forms. [12] The pathogenesis of PPV remains elusive. In the year 1993, Happle proposed a genetic phenomenon called "twin spotting" to explain the etiopathogenesis. In their opinion, PPV may be a result of genetic mosaicism and allelic mutation. [13]
In the present case, clinical findings of PWS along with ocular melanosis and neurological involvement were consistent with the traditional PPV type IIB or its counterpart of phakomatosis cesioflammea. Radiological assessment showed brain atrophy and calcification, muscular and soft tissue and bony hypertrophy of the left upper limb along with venous varicosities, which were consistent with the diagnosis of overlap between Sturge-Weber and Klippel-Trenaunay syndromes. These interesting combinations of clinical and radiological findings have been extremely rarely reported in the literature. [14],[15],[16]
Phakomatosis pigmentovascularis has also been reported to be associated with temporal alopecia, malignant colon polyposis, scoliosis and leg length discrepancy, hypoplastic larynx and subglottic stenosis, multiple granular cell tumor, selective IgA deficiency, iris hamartomas, and generalized vitiligo. [17],[18],[19],[20],[21],[22] However, our patient did not have any such abnormalities.
In this report, we emphasize the importance of complete clinical and radiological evaluation of cases that present with PPV. This helps in identifying any underlying condition early in the course of the disease, which may determine the outcome of this condition.
| 1. |
Mahroughan M, Mehregan AH, Mehregan DA. Phakomatosis pigmentovascularis: Report of a case. Pediatr Dermatol 1996;13:36-8.
[Google Scholar]
|
| 2. |
Hasegawa Y, Yasuhara M. Phakomatosis pigmentovascularis type IVa. Arch Dermatol 1985;121:651-5.
[Google Scholar]
|
| 3. |
Cull RE, White IR. The nervous system. In: Munro J, Edwards C, editors. Macleod's Clinical Examination. 9 th ed. Edinburgh: Churchill Livingstone; 1995. p. 201-56.
th ed. Edinburgh: Churchill Livingstone; 1995. p. 201-56.'>[Google Scholar]
|
| 4. |
Leung AK, Lowry RB, Mitchell I, Martin S, Cooper DM. Klippel-Trenaunay and Sturge-Weber syndrome with extensive mongolian spots, hypoplastic larynx and subglottic stenosis. Clin Exp Dermatol 1988;13:128-32.
[Google Scholar]
|
| 5. |
Uysal G, Güven A, Ozhan B, Oztürk MH, Mutluay AH, Tulunay O. Phakomatosis pigmentovascularis with Sturge-Weber syndrome: A case report. J Dermatol 2000;27:467-70.
[Google Scholar]
|
| 6. |
Van Gysel D, Oranje AP, Stroink H, Simonsz HJ. Phakomatosis pigmentovascularis. Pediatr Dermatol 1996;13:33-5.
[Google Scholar]
|
| 7. |
Du LC, Delaporte E, Catteau B, Destee A, Piette F. Phakomatosis pigmentovascularis type II. Eur J Dermatol 1998;8:569-72.
[Google Scholar]
|
| 8. |
Bielsa I, Paradelo C, Riberta M, Ferrándiz C. Generalized nevus spilus and nevus anemicus in a patient with a primary lymphedema: A new type of phakomatosis pigmentovascularis? Pediatr Dermatol 1998;15:293-5.
[Google Scholar]
|
| 9. |
Di Landro A, Tadini GL, Marchesi L, Cainelli T. Phakomatosis pigmentovascularis: A new case with renal angiomas and some consideration about the classification. Pediatr Dermatol 1999;16:25-30.
[Google Scholar]
|
| 10. |
Vidaurri-de la Cruz H, Tamayo-Sanchez L, Duran-McKinster C, Orozco-Covarrubias ML, Ruiz-Maldonado R. Phacomatosis pigmentovascularis IIA and IIB: Clinical findings in 24 patients. J Dermatol 2003;30:381-8.
[Google Scholar]
|
| 11. |
Ruiz-Maldonado R, Tamayo L, Laterza AM, Brawn G, Lopez A. Phacomatosis pigmentovascularis: A new syndrome? Report of four cases. Pediatr Dermatol 1987;4:189-96.
[Google Scholar]
|
| 12. |
Happle R. Phacomatosis pigmentovascularis revisited and reclassified. Arch Dermatol 2005;141:385-8.
[Google Scholar]
|
| 13. |
Happle R. Allelic somatic mutations may explain vascular twin nevi. Hum Genet 1991;86:321-2.
[Google Scholar]
|
| 14. |
Finklea LB, Mohr MR, Warthan MM, Darrow DH, Williams JV. Two reports of phacomatosis pigmentovascularis type IIb, one in association with Sturge-Weber syndrome and Klippel-Trenaunay syndrome. Pediatr Dermatol 2010;27:303-5.
[Google Scholar]
|
| 15. |
Saricaoðlu MS, Güven D, Karakurt A, Sengun A, Ziraman I. An unusual case of Sturge-Weber syndrome in association with phakomatosis pigmentovascularis and Klippel-Trenaunay-Weber syndrome. Retina 2002;22:368-71.
[Google Scholar]
|
| 16. |
Chhajed M, Pandit S, Dhawan N, Jain A. Klippel-Trenaunay and Sturge-Weber overlap syndrome with phakomatosis pigmentovascularis. J Pediatr Neurosci 2010;5:138-40.
[Google Scholar]
|
| 17. |
Kikuchi I, Okazaki M. Congenital temporal alopecia in phakomatosis pigmentovascularis. J Dermatol 1982;9:485-7.
[Google Scholar]
|
| 18. |
Horio T, Ogawa M. Pigmentovascular nevus. Arch Dermatol 1973;107:463-4.
[Google Scholar]
|
| 19. |
Guiglia MC, Prendiville JS. Multiple granular cell tumors associated with giant speckled lentiginous nevus and nevus flammeus in a child. J Am Acad Dermatol 1991;24:359-63.
[Google Scholar]
|
| 20. |
de Luna ML, Barquin MA, Casas IG, Sidelsky S. Phakomatosis pigmentovascularis with a selective IgA deficiency. Pediatr Dermatol 1995;12:159-63.
[Google Scholar]
|
| 21. |
Gilliam AC, Ragge NK, Perez MI, Bolognia JL. Phakomatosis pigmentovascularis type IIb with iris mammillations. Arch Dermatol 1993;129:340-2.
[Google Scholar]
|
| 22. |
Kim YC, Park HJ, Cinn YW. Phakomatosis pigmentovascularis type IIa with generalized vitiligo. Br J Dermatol 2002;147:1028-9.
[Google Scholar]
|
Fulltext Views
5,193
PDF downloads
2,389





![[Figure - 1]](#fig_ijdvl_2014_80_1_51_125507_f1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2014_80_1_51_125507_f2.jpg){kind=link}
![[Figure - 3]](#fig_ijdvl_2014_80_1_51_125507_f3.jpg){kind=link}
![[Figure - 4]](#fig_ijdvl_2014_80_1_51_125507_f4.jpg){kind=link}