Translate this page into:
Tonsillar fat herniation: A novel finding in Goltz syndrome
Correspondence Address:
Zhimiao Lin
Department of Dermatology, Peking University First Hospital, Beijing 100034
China
| How to cite this article: Wang Z, Wang H, Lin Z. Tonsillar fat herniation: A novel finding in Goltz syndrome. Indian J Dermatol Venereol Leprol 2020;86:691-693 |
Sir,
Goltz syndrome (focal dermal hypoplasia; OMIM #305600) is a rare multisystem genetic disorder affecting skin, eyes, teeth, nails and the skeleton.[1] Subcutaneous fat herniation is a hallmark of Goltz syndrome. However, we were unable to find any previous reports of tonsillar fat herniation. We, herein, report the first case of tonsillar fat herniation in Goltz syndrome.
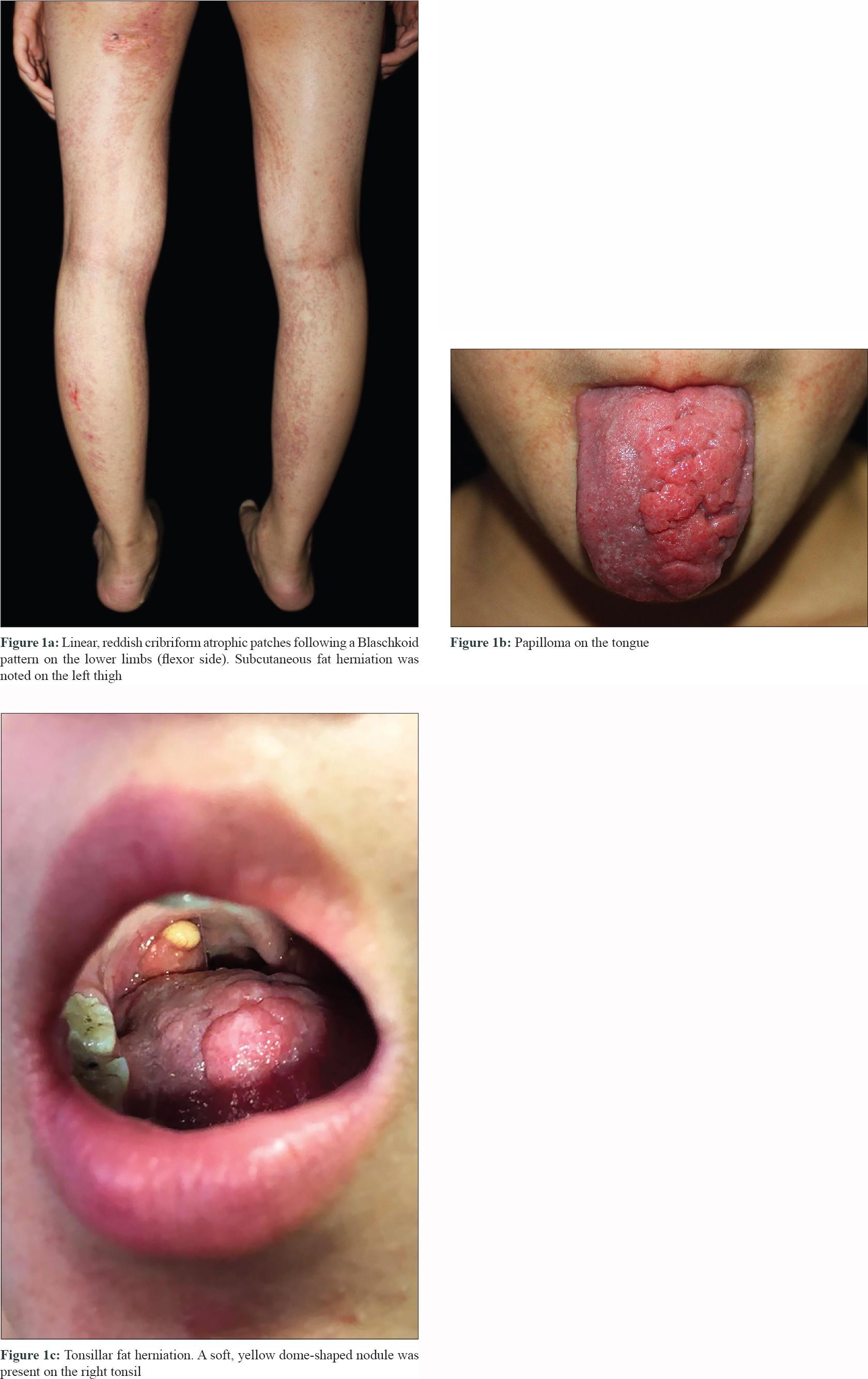
A 12-year-old girl was referred to Peking UniversityFirst Hospital in Beijing, China. She presented with streaks of hyperpigmentation and skin atrophy with telangiectasia following a blaschkoid pattern soon after birth. Physical examination revealed linear, slightly depressed, reddish cribriform atrophic patches generalized distributed on her extremities [Figure - 1]a, trunk and face, and linear hyperpigmented patches on her dorsal trunk. Subcutaneous fat herniation was noted on the left thigh [Figure - 1]a. Streaks of papillomatous hyperkeratosis were found on her hands and feet with ridged, dysplastic nails and brachydactyly of the left fourth toe. Examination of oral cavity showed oligodontia, enamel hypoplasia and multiple papillomatous growths on her tongue [Figure - 1]b, hard palate and left tonsil. Remarkably, a soft, yellow, dome-shaped nodule, which was reminiscent of fat herniation, was present on her right tonsil [Figure - 1]c. Family history was negative. Histopathology of the atrophic skin lesion revealed decreased thickness of dermis and absence of adnexal structures [Figure - 2]. To identify the underlying genetic defect, genomic DNA was extracted from peripheral blood leukocytes after obtaining written consent from the parents. Although Sanger sequencing of the exons and flanking regions of PORCN gene failed to reveal any pathogenic mutation, a clinical diagnosis of Goltz syndrome was made based on the characteristic clinical and histopathological features of the patient.
 |
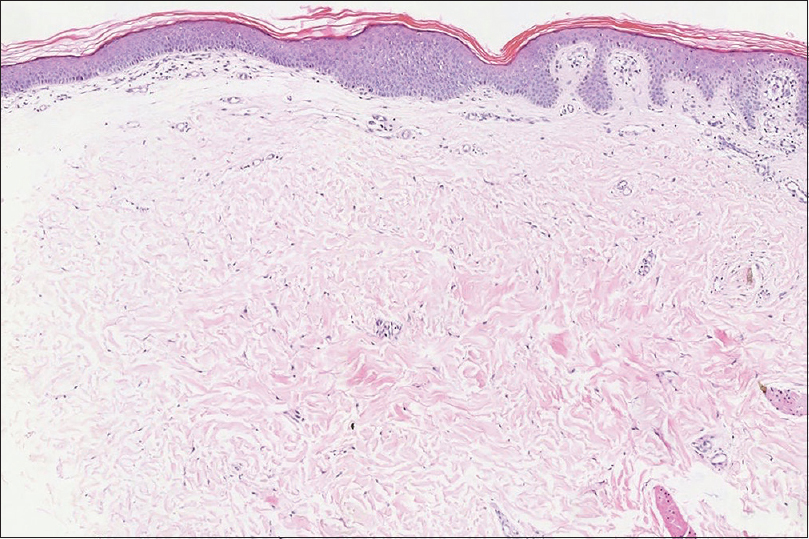 |
| Figure 2: Decreased thickness of dermis, absence of adnexal structures, hyperkeratosis and acanthosis of epidermis (H and E, ×100) |
Goltz syndrome is a rare X-linked dominant genodermatosis affecting multiple tissues and organs of meso-ectodermal origin. It is characterized by pathognomonic skin manifestations and variable abnormalities that mainly involve eyes, teeth, nails and skeleton.[1] Skin involvement is characterized by congenital patchy skin aplasia and pigmentary changes along the Blaschko's lines, often accompanied by telangiectasia. Papillomas may appear in any location, especially the orifices and acral sites. Another hallmark of Goltz syndrome is subcutaneous fat herniation, which is manifested as soft, yellow nodules usually found on the atrophic skin. Interestingly, this is the first reported case of a patient with fat herniation on the tonsil. The tonsil derives from mesoderm, both herniation of fat into a dysplastic tonsil and ectopic aggregation of fat within the tonsil can explain this manifestation. Imaging tests such as magnetic resonance imaging are expected to differentiate these two conditions, since herniation typically appears as a pedunculated mass. A biopsy on this site may also aid by clarifying the development of the tonsil and the distribution of fat. However, they were declined by the patient. Histopathological findings of Goltz syndrome range from a classic pattern of replacement of the dermis with fat to a less recognized pattern of increased capillaries in the papillary dermis and adipocytes within/below the thinned dermis.[2]
Goltz syndrome is caused by loss-of-function mutations of PORCN gene, which is mapped to chromosome Xp11.23.[1] Either large deletion of PORCN or post-zygotic mosaicism may explain the negative result of genetic testing in this case. PORCN encodes an O-acyltransferase which is involved in the palmitoylation and secretion of Wnt signaling proteins. Wnt signaling is crucial to meso-ectodermal tissue development, especially for fibroblast proliferation and osteogenesis, which explains the dermal hypoplasia and skeleton anomalies observed in Goltz syndrome.[3]
Bostwick et al. have proposed a set of clinical diagnostic criteria for Goltz syndrome.[1] They stated that diagnosis should be made if a patient displays three or more of five characteristic skin manifestations and at least one of five characteristic limb malformations. The patient in this case presented with all the five characteristic skin findings. Although she did not have any characteristic limb malformations, brachydactyly was detected, indicating a skeletal involvement. In fact, the incidence of brachydactyly in Goltz syndrome is higher than that of transverse limb defects and oligodactyly.[4] Probably due to higher specificity, Bostwick et al. incorporated the latter two, rather than brachydactyly, in the criteria. However, due to random X-inactivation, mosaicism and the mutation itself, Goltz syndrome actually has a wide clinical spectrum from very subtle manifestations to severe findings. There are even reports of patients without skin manifestations.[5] Therefore the full-blown manifestations might be present only in typical cases, while for atypical ones, biopsy and genetic testing can be used to aid diagnosis. The differential diagnosis includes other disorders with a blaschkoid distribution of skin lesions, such as incontinentia pigmenti and MIDAS (microphtalmia, dermal aplasia, and sclerocornea) syndrome. Treatment is mainly supportive. No treatment was planned for the skin lesions of the patient at the time since no complaint was reported.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form, the legal guardians have given their consent for images and other clinical information to be reported in the journal. The guardians understand that names and initials will not be published and due efforts will be made to conceal identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
This study was supported financially by Peking University Medicine Fund of Fostering Young Scholar's Scientific and Technological Innovation (grant no. BMU2020PY006).
Conflicts of interest
There are no conflicts of interest.
| 1. |
Bostwick B, Fang P, Patel A, Sutton VR. Phenotypic and molecular characterization of focal dermal hypoplasia in 18 individuals. Am J Med Genet C Semin Med Genet 2016;172C: 9-20.
[Google Scholar]
|
| 2. |
Ko CJ, Antaya RJ, Zubek A, Craiglow B, Damsky W, Galan A, et al. Revisiting histopathologic findings in Goltz syndrome. J Cutan Pathol 2016;43:418-21.
[Google Scholar]
|
| 3. |
Barrott JJ, Cash GM, Smith AP, Barrow JR, Murtaugh LC. Deletion of mouse Porcn blocks Wnt ligand secretion and reveals an ectodermal etiology of human focal dermal hypoplasia/Goltz syndrome. Proc Natl Acad Sci U S A 2011;108:12752-7.
[Google Scholar]
|
| 4. |
Smith A, Hunt TR 3rd. The orthopedic characterization of Goltz syndrome. Am J Med Genet C Semin Med Genet 2016;172C: 41-3.
[Google Scholar]
|
| 5. |
Madan S, Liu W, Lu JT, Sutton VR, Toth B, Joe P, et al. A non-mosaic PORCN mutation in a male with severe congenital anomalies overlapping focal dermal hypoplasia. Mol Genet Metab Rep 2017;12:57-61.
[Google Scholar]
|
Fulltext Views
3,267
PDF downloads
1,451





![[Figure - 1]](#fig_ijdvl_2020_86_6_691_295633_f1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2020_86_6_691_295633_f2.jpg){kind=link}