
Translate this page into:
Unveiling a novel gene mutation in neonatal ichthyosis-sclerosing cholangitis syndrome with progressive liver disease

Corresponding author: Dr. Neetu Bhari, Department of Dermatology & Venereology, All India Institute of Medical Sciences, New Delhi, India. drntbhari@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Danish M, Choudhary R, Bhari N. Unveiling a novel gene mutation in neonatal ichthyosis-sclerosing cholangitis syndrome with progressive liver disease. Indian J Dermatol Venereol Leprol. doi: 10.25259/IJDVL_436_2024
Dear Editor,
A 4-year-old boy, born to third-degree consanguineous parents, presented with generalised dry, scaly, and itchy skin since birth, alongwith thinning of hair on the scalp and eyebrows. At one-month-old, he developed jaundice, high-coloured urine, pale stools, and palpable hepatomegaly. Laboratory tests revealed serum total bilirubin levels of 15.1 mg/dl (0.2–1 mg/dl), conjugated bilirubin levels of 11.5 mg/dl (0.2–0.3 mg/dl), aspartate aminotransferase (AST) of 150 IU/L (10–40 IU/L), alanine aminotransferase (ALT) of 81 IU/L (8–33 U/L), gamma-glutamyl transferase (GGT) of 81 IU/L (5–55 IU/L), alkaline phosphatase (ALP) of 597 IU/L (70–400 U/L) and serum bile acids of 78 µmol/L (<10 µmol/L). Ultrasound of the abdomen showed no significant abnormalities. Liver biopsy revealed preserved lobular architecture, mixed inflammatory infiltrates around bile ducts, mild hepatocellular cholestasis, and focal giant cells in hepatocytes. Hepato-porto-enterostomy at the age of one month improved his liver function tests (SGOT/SGPT: 64/61 IU/L, total/direct bilirubin: 2.7/1.9 g/dl from 150/81 and 15.1/11.5 g/dl). At eight months, the child developed generalised pruritus, which responded with varying doses of ursodeoxycholic acid (UDCA) and levocetirizine. After that, he continued to develop itching with partial response to levocetirizine and UDCA in varying doses. Gradually, he started developing brownish scales all over his body and was advised to use emollients. Examination revealed generalised dry brownish hyperpigmented scales [Figure 1a] along with sparsening of hair over the scalp and eyebrows [Figure 1b], hypodontia, and enamel defects [Figure 2].
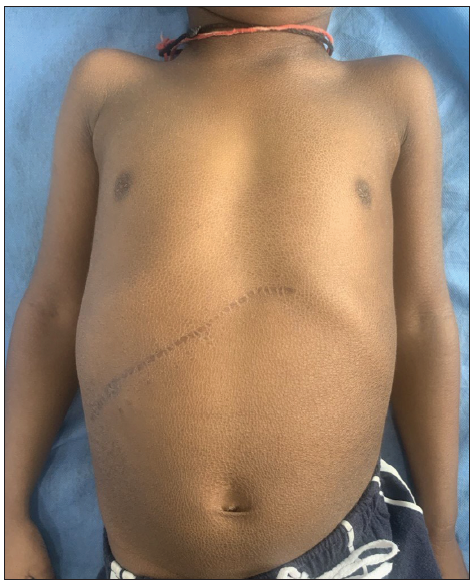
- Diffuse brownish hyperpigmented ichthyotic scales over the trunk with the linear hyperpigmented scar of previous surgery in the right subcostal area.
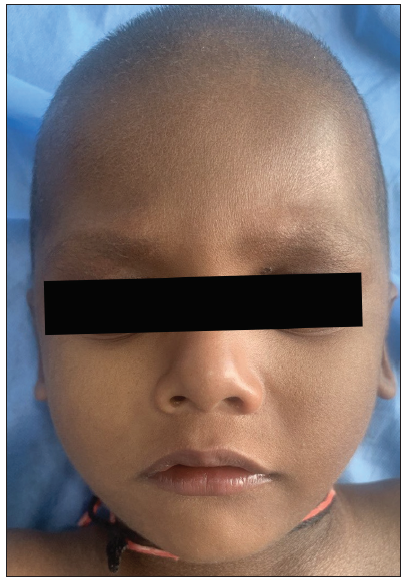
- Diffuse sparsening of hair over scalp and eyebrows along with mild ichthyosis over the face.
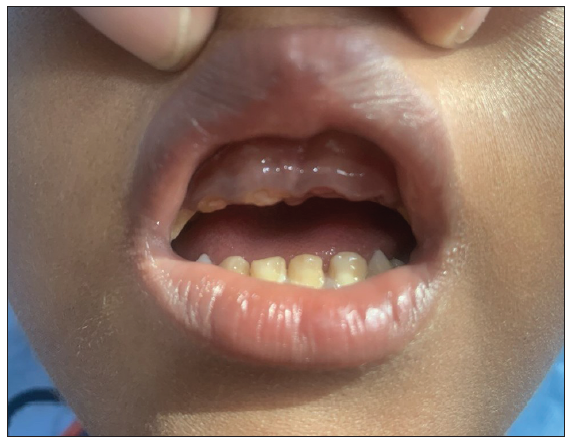
- Dental enamel defect with hypodontia.
On the evaluation of liver function, total and direct bilirubin were 7 g/dl and 5.2 g/dl, respectively, SGOT and SGPT were 86 IU/L and 64 IU/L and serum albumin was 2 g/dl (3.5–5 g/dl). Repeat ultrasonography revealed chronic liver disease changes (coarse echoes with irregular texture and left lobe hypertrophy, with no obvious space-occupying lesions or intrahepatic biliary radicle dilatation (IHBRD). A diagnosis of neonatal ichthyosis-sclerosing cholangitis (NISCH) syndrome was confirmed through the next-generation sequencing which revealed a homozygous mutation in the CLDN1 gene (exon 1 variant c.191G>T, p.Cys64Phe, Depth: 182x). According to the American College of Medical Genetics recommendations, this mutation is of uncertain significance. This variant has not been reported in 1000 genomes, clinvar, and gnom AD databases, and has a minor allele frequency of 0.003% in the internal database of MedGenome Labs Ltd, Bangalore, India. In silico prediction of the variant are probably damaged by Polyphen-2 (HumDiv) and damaged by SIFT (sorting intolerant from tolerant), LRT (Likelihood Ratio Test), and MutationTaster2. Due to financial constraints, genetic testing for the parents was not done. The child was referred to the gastroenterology department for derangement of liver parameters, the dose of UDCA was hiked and the application of white soft paraffin was advised.
NISCH syndrome is a rare autosomal recessive disorder characterised by neonatal ichthyosis and sclerosing cholangitis. It is inherited as an autosomal recessive disorder. Due to the extreme rarity of the disorder, the exact prevalence is unknown; however, 21 cases have been reported in the literature until 2023. Alopecia, hypotrichosis, and ichthyosis have been reported in all cases. Dentition abnormalities, such as enamel hypoplasia and hypodontia, are seen in around 64% of patients.1 Liver involvement is heterogeneous ranging from no clinically evident disease to liver cirrhosis. Liver sonography usually appears normal, while liver biopsy reveals cholestasis, portal fibrosis, ductular proliferation, and ductular paucity.1 Our patient had a classic presentation in the form of generalised mild ichthyosis with itching, sparsening of hair, dental abnormalities, and neonatal cholangitis leading to liver cirrhosis. NISCH results from homozygous CLDN1 gene mutations which encodes for claudin-1, vital for tight junctions, which are essential for skin and liver cell connections. Various mutations have been reported in CLDN1 gene exons, including exon 1 (c.200_201delTT), the Moroccan mutation2; homozygous deletion in the nucleotide of exon 1 (p.Val66_Phe67insTer); exon 2 (c.358delG) in the Swiss population3; (c.181C>T, p.Gln61X) in a Turkish family4; and (c.578C>A, p.Tyr159Ter) in the Iranian population5 [Table 1]. The ‘Swiss’ and ‘Turkish’ mutations show a better prognosis, while the ‘Moroccan’ mutation leads to diverse phenotypes, ranging from persistent liver disease to no liver involvement, making it difficult to predict prognosis. Our patient had a novel mutation (c.191G>T, p.Cys64Phe) in exon 1, previously unreported, and experienced persistent ichthyosis and progressive liver disease, resulting in cirrhosis by the age of four. The differential diagnosis includes other inherited disorders associated with liver involvement and ichthyosis [Table 2]. The prognosis of NISCH remains unclear due to the limited number of published reports. Some genetic variants associated with NISCH have exhibited varying degrees of extracutaneous involvement, including liver abnormalities. However, more studies are needed to better understand the prognosis and broader implications of this condition.
| Type of mutation variant based on geographic location | Gene | Location | Variant | Prognosis |
|---|---|---|---|---|
| Moroccan2 | CLDN1 | Exon 1 | (c.200_201delTT) | Variable; persistent liver disease to no liver involvement |
| Swiss3 | CLDN1 | Exon 1 | (p.Val66_Phe67insTer) | Regression of cholestasis |
| CLDN1 | Exon 2 | (c.358delG) | Regression of cholestasis | |
| Turkish4 | CLDN1 | Exon 1 | (c.181C>T, p.Gln61X) |
Neonatal jaundice recovering by 6–12 months Ichthyosis and hypotrichosis persisting |
| Iranian5 | CLDN1 | Exon 1 | (c.578C>A, p.Tyr159Ter) | Follow-up findings not available |
| Indian6 | CLDN1 | Exon 1 | (p.Tyr47Ter) | Cholestasis resolved in infancy |
| Current report | CLDN1 | Exon 1 | (c.191G>T, p.Cys64Phe) | Progressing to chronic liver disease |
| Disorder | Mutation | Clinical features |
|---|---|---|
| MEDNIK Syndrome | AP1S1 gene |
Mental retardation, enteropathy, deafness, peripheral neuropathy, ichthyosis, keratoderma Neonatal cholestasis |
| ARC Syndrome | VPS33B or VIPAR gene |
Arthrogryposis, renal dysfunction, cholestasis Severe developmental delay |
| Zellweger Spectrum Disorders | PEX gene |
Ichthyosis, liver dysfunction, hypotonia Neurological impairment |
| Type 2 Gaucher’s Disease | GBA gene | Neonatal cholestasis, hepatosplenomegaly, ichthyosis, neurological decline |
MEDNIK: Mental retardation, deafness, neuropathy, ichthyosis, and keratodermia, ARC: Arthrogryposis-renal dysfunction-cholestasis.
In conclusion, we would like to highlight that genetic testing is essential for accurate diagnosis and that the specific CLDN1 gene mutation can influence the severity of liver disease. Understanding the genetic basis of NISCH can help predict disease progression and guide appropriate interventions in affected individuals.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of AI-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
References
- Neonatal ichthyosis-sclerosing cholangitis syndrome: Report of a novel mutation and a review of the literature. Clin Dysmorphol. 2023;32:88-91.
- [CrossRef] [PubMed] [Google Scholar]
- Homozygosity mapping of a locus for a novel syndromic ichthyosis to chromosome 3q27-q28. J Invest Dermatol. 2002;119:70-76.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Confirmation of the origin of NISCH syndrome. Hum Mutat. 2006;27:408-10.
- [CrossRef] [PubMed] [Google Scholar]
- Novel mutation in the CLDN1 gene in a Turkish family with neonatal ichthyosis sclerosing cholangitis (NISCH) syndrome. Br J Dermatol. 2014;170:976-78.
- [CrossRef] [PubMed] [Google Scholar]
- Gene-targeted next-generation sequencing identifies a novel CLDN1 mutation in a consanguineous family with NISCH syndrome. Am J Gastroenterol. 2017;112:396-98.
- [CrossRef] [PubMed] [Google Scholar]
- Neonatal Ichthyosis and sclerosing cholangitis (NISCH) syndrome with a novel Claudin-1 (CLDN1) mutation: A report from India. Indian J Dermatol Venereol Leprol. 2023;19:1-3.
- [CrossRef] [Google Scholar]




