Translate this page into:
Vulvar ulcer as a presentation of systemic langerhans cell histiocytosis
Correspondence Address:
Nina A Madnani
Department of Dermatology, P.D.Hinduja Hospital and MRC, Mumbai - 400 016
India
| How to cite this article: Madnani NA, Khan KJ. Vulvar ulcer as a presentation of systemic langerhans cell histiocytosis. Indian J Dermatol Venereol Leprol 2011;77:177-179 |
Abstract
We report a 38-year-old housewife with systemic Langerhans cell histiocytosis (LCH) presenting as a chronic vulvar and peri-anal ulcer. She had systemic involvement in the form of diabetes insipidus and bone "hot-spots". She responded favorably to etoposide, 6-mercaptopurine, and systemic steroids, and has been in remission since 10 years. Chronic vulvar ulcers not responding to routine therapy should not be neglected and need to be biopsied repeatedly to come to a specific diagnosis. The vulvar ulcer in our case provided a vital clue to a systemic LCH, with a successful outcome.Introduction
Chronic vulvar ulcers may occur as a primary disease or a manifestation of a systemic process with causes ranging from Behcet′s disease, cutaneous tuberculosis, amebiasis, neoplasias, Crohn′s disease, and even Langerhans cell histiocytosis (LCH) to name a few. We present a case of a housewife who presented with a chronic vulvar ulcer as a part of a systemic spectrum of LCH.
Case Report
In 1999, a 38-year-old housewife presented with a 5 year chronic, painful, vulvar ulcer. Within a year of her last child birth, 12 years prior, she developed amenorrhea and over the next 3-4 years, heat intolerance and excessive thirst, frequent headaches, and mood swings. Over the last 3 years, she suffered severe leg pains causing her to limp. She had never suffered from otorrhea, dyspnea, diarrhea, or weight fluctuation. Her previous investigations revealed a consistently raised ESR, a normal ultrasound of the pelvis, and a positive TB IgG antigen titer. Multiple past biopsies from the ulcer were inconclusive. Multiple antibiotic courses including a 6 month course of anti-tuberculous treatment, and anti-amebic medications gave no relief.
Examination of the vulva showed a well-defined, tender, fleshy, indurated ulcer on the left labia minora, extending backward to the perianal area and into the natal cleft [Figure - 1]. Clinical differential diagnosis included vulvar tuberculosis, cutaneous amebiasis, vulvar Crohn′s disease, and neoplasia. Her mouth had few teeth with extensive gum resorption [[Figure - 2]a]. Systemic examination was unremarkable. Histopathology of the ulcer edge biopsy showed an ulcerated epidermis with diffuse infiltration of the dermis with mononuclear cells, most of which were large histiocytic cells with abundant cytoplasm and an indented nucleus [[Figure - 3]a and b]. These were suggestive of Langerhans cells and confirmed with special staining with S-100 [[Figure - 3]c]. A bone scan revealed multiple "hot-spots" in various parts of the skeleton [[Figure - 2]b]. Routine hematological investigations including liver and renal function tests were within normal limits. Levels of FSH 1.6 mIU/ml (36.6-168.8 post-menopausal) and LH 0.49 mIU/ml (14.4-62.2 post-menopausal) were indicative of premature ovarian failure. Urine osmolality of 70 mOs/kg (560-850) and water deprivation test confirmed a diabetes insipidus (DI). A computerized tomography (CT) of the abdomen showed diffuse infiltration of the liver, and a Magnetic resonance imaging (MRI) of the brain showed an enhancing isointense mass involving the floor of the third ventricle, tuber cinereum, mamillary bodies, and posterior part of the optic chiasma. Her bone marrow was normal. In view of her clinical and investigational findings, we labeled her disease as multi-system LCH.
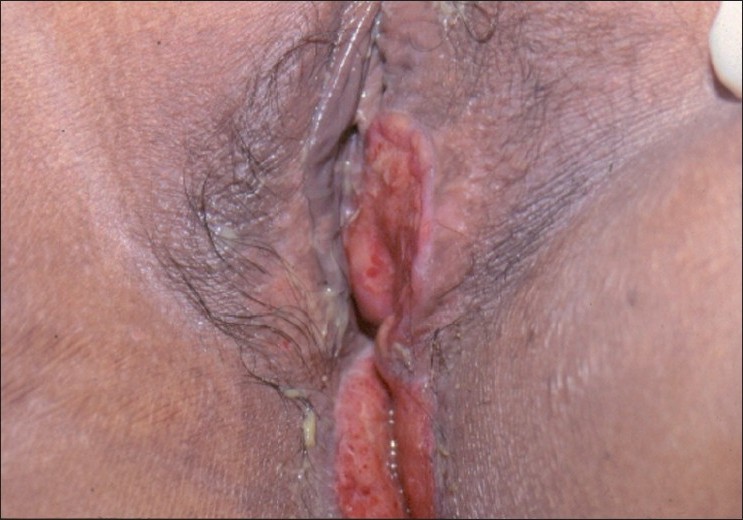 |
| Figure 1: Fleshy ulcer extending from the left labia minora backward to peri-anal area |
 |
| Figure 2: (a) Radiography of jaw showing gum resorption and "floating" teeth. (b) Bone scan showing multiple "hot spots" in the skeleton |
 |
| Figure 3: (a) Hyperplastic, ulcerated epidermis with diffuse dermal inflammatory infiltrate (H and E, �40). (b) Numerous histiocytes with large indented nucleus and abundant cytoplasm seen (H and E, ×200). (c) S-100 stain +ve histiocytes seen scattered in the dermis (H and E, ×200) |
Treatment was instituted with etoposide 50 mg daily in a cyclical regime of 3 weeks on and 1 week off, to a total dose of 4950 mg, followed by 6-mercaptopurine 100 mg daily with prednisolone 40 mg on alternate days. The steroids were tapered off over a year. Her diabetes insipidus was controlled with desmopressin puffs. The ulcer healed in 3 months with disappearance of the bone pains [Figure - 4]. The patient has been in clinical remission for the last 10 years.
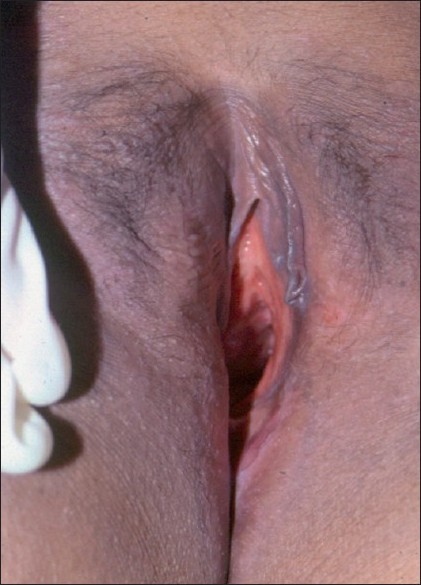 |
| Figure 4: Healed labial and peri-anal ulcer |
Discussion
LCH is part of a larger group of syndromes described as histiocytoses. The disease may involve single or multiple systems including bones, skin, reticulo-endothelial system, nervous system, and endocrine glands with bone being the most common. [1] The Histiocyte Society, classifies LCH as Single System LCH (SS-LCH) or Multisystem LCH (MS-LCH) depending on the extent and localization of disease (organ/system involvement). [2] The disease results from uncontrolled proliferation of Langerhans cells, the cause of which is unknown but is now postulated to be due to viral infections which cause abnormal cytokine release resulting in clonal proliferation. [3]
The involvement of the lower genital tract in LCH is not unexpected as Langerhans cells in the genital mucosa transformation zone perform the important function of immune surveillance. This function is directly influenced by external factors such as human papilloma virus and human herpes virus infections. [4] The lesions may present as an area of induration, pruritic rash, papules or ulceration, and fissuring. Involvement of the oral mucosa with gingival nodules or ulceration often accompanies the genital involvement. The skull, long bones, and flat bones are commonly involved. A bone scan of our patient revealed multiple "hot-spots" in the calvarium, jaw, and long bones indicating extensive bone involvement. Diabetes insipidus (DI) is seen in more than 50% of patients with skull involvement [5] and can be confirmed by urine osmolality after water deprivation. Our patient tested positive and is well controlled with vasopressin puffs. Grois et al, concluded in their study that patients with MS-LCH with oro-facial involvement had a higher risk of developing DI. [6]
The histopathology remains the same across the spectrum of disease with infiltration by characteristic Langerhans cells. These cells are identified by their relatively large size having a distinct, folded, or lobulated, often kidney-shaped nucleus, with a higher nucleus to cytoplasmic ratio than normal macrophages. Special stains like S100, CD1a and langerin are confirmatory and demonstrating Birbeck granules on electron microscopy is no longer the gold standard. Due to economic constraints, the electron microscopy for Birbeck granules and CD1a staining were not performed. Testing for langerin is not available in India.
Treatment is dictated by the severity of disease and organ system(s) involved.Possibility of spontaneous remission should caution against aggressive treatment options early in the disease. Skin involvement can be treated with super potent topical steroids, 20% topical nitrogen mustard, PUVA, IFN-A, and thalidomide. [7] Isolated vulvar involvement has been managed with surgical excision with excellent results. [8] Systemic therapy is indicated in SS-LCH with "CNS-risk" lesions, SS-LCH with multifocal bone disease (MFB), SS-LCH with "special-site" lesions, and MS-LCH with/without involvement of "risk organs". The Histiocyte Society recommends an initial 6 week course of vinblastine and prednisone, and reassessment in 6 weeks, followed by maintenance of 3 week pulses of vinblastine and prednisolone and daily 6-mercaptopurine for total of 12 months. Our patient responded to etoposide, 6-MP, and prednisolone. She is in remission till today.
The above case report highlights the need for an aggressive work up of every persistent non-healing vulvar ulcer, with multiple biopsies performed, to determine the etiology.
| 1. |
X-type Histiocytoses (Macrophage/Monocyte Disorders) In: James WD, Berger TG, Elston DM, editors. Andrews′ Diseases of the skin. 10 th ed. Canada: Saunders Elsevier; 2006. p. 722.
[Google Scholar]
|
| 2. |
The Histiocyte Society. Available from: http://www.histiocytesociety.org . [Last accessed on 2009 Sep 12].
[Google Scholar]
|
| 3. |
Csire M, Mikala G, Jákó J, Masszi T, Jánosi J, Dolgos J, et al. Persistent long-term human herpesvirus 6 (HHV-6) infection in a patient with langerhans cell histiocytosis. Pathol Oncol Res 2007;13:157-60.
[Google Scholar]
|
| 4. |
Pan Z, Sharma S, Sharma P. Primary Langerhans cell histiocytosis of the vulva: Report of a case and brief review of the literature. Indian J Pathol Microbiol 2009;52:65-8.
[Google Scholar]
|
| 5. |
Caputo R, Gelmetti C. Langerhans cell histiocytosis. In: Wolff K, Goldsmith LA, Katz SI, Gilchrest BA, Paller AS, Leffell DJ, editors. Fitzpatrick′s Dermatology in General Medicine. 7 th ed. USA: McGraw Hill; 2008. p. 1419.
[Google Scholar]
|
| 6. |
Grois N, Pötschger U, Prosch H, Minkov M, Arico M, Braier J, et al. Risk factors for diabetes insipidus in Langerhans cell histiocytosis. Pediatr Blood Cancer 2006;46:228-33.
[Google Scholar]
|
| 7. |
Broekaert SM, Metzler G, Burgdorf W, Röcken M, Schaller M. Multisystem Langerhans cell histiocytosis: Successful treatment with thalidomide. Am J Clin Dermatol 2007;8:311-4.
[Google Scholar]
|
| 8. |
Ishigaki H, Hatta N, Yamada M, Orito H, Takehara K. Localised vulva Langerhans cell histiocytosis. Eur J Dermatol 2004;14:412-4.
[Google Scholar]
|
Fulltext Views
3,075
PDF downloads
2,569





![[Figure - 1]](#fig_ijdvl_2011_77_2_177_77458_f1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2011_77_2_177_77458_f2.jpg){kind=link}
![[Figure - 3]](#fig_ijdvl_2011_77_2_177_77458_f3.jpg){kind=link}
![[Figure - 4]](#fig_ijdvl_2011_77_2_177_77458_f4.jpg){kind=link}