Translate this page into:
Waardenburg syndrome
2 Department of Ophthalmology, Government Medical College, Patiala - 147 001, Punjab, India
Correspondence Address:
Sunita Tagra
55, Surjit Nagar, Kapurthala Road, Jalandhar City, Punjab - 144 002
India
| How to cite this article: Tagra S, Talwar AK, Walia RS, Sidhu P. Waardenburg syndrome. Indian J Dermatol Venereol Leprol 2006;72:326 |
Abstract
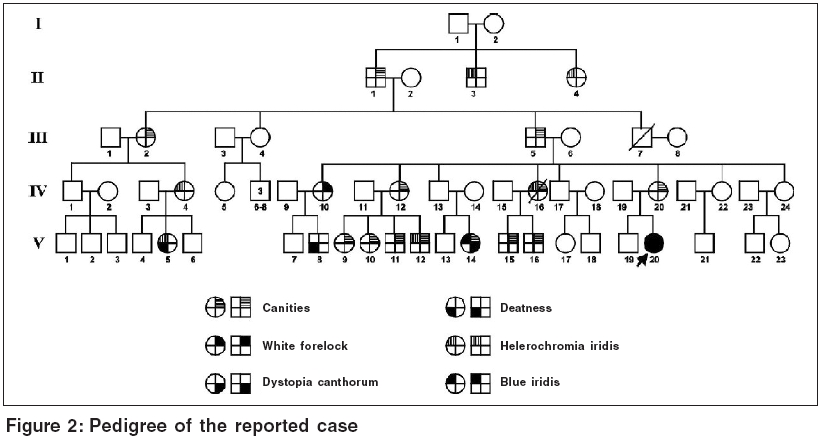
Waardenburg syndrome is a rare inherited and genetically heterogenous disorder of neural crest cell development. Four distinct subtypes showing marked interfamilial and intrafamilial variability have been described. We report a girl showing constellation of congenital hearing impairment with 110 dB and 105 dB loss in right and left ear respectively, hypoplastic blue iridis, white forelock, dystopia canthorum and broad nasal root. Other affected relatives of the family, with variable features of the syndrome, have been depicted in the pedigree.
 |
|
 |
|
Introduction
Waardenburg syndrome (WS) is named after P. J. Waardenburg, a Dutch ophthalmologist, who in 1951 described a syndrome with six characteristic features; lateral displacement of the medial canthi and lacrimal punctae, broad and high nasal root, hypertrichosis of medial part of eyebrows, partial or total heterochromia iridis, white forelock and congenital deaf-mutism. Since then, four subtypes (I to IV) with variable penetrance and gene expression of different clinical features have been described.[1] Of these WS I and II are autosomal dominant in most of cases. WS III (Klein-Waardenburg syndrome) is usually sporadic but when it occurs in families, inheritance is autosomal dominant. Type IV (Shah-Waardenburg syndrome) is probably autosomal recessive.[1],[2],[3] WS accounts for about 2 to 5% cases of congenital deafness.[3] In about 20% of cases, syndrome is expressed in incomplete form.[1]
As per the diagnostic criteria proposed by Waardenburg consortium [Table - 1], for diagnosing WS I, two major or one major plus two minor criteria should be present.[4] WS III is similar to type I with additional musculoskeletal abnormalities. WS IV is associated with Hirschsprung disease.[1],[2]
WS II is characterized by more frequent occurrence of sensorineural hearing loss and heterochromia iridis but absence of dystopia canthorum. Liu et al[5] have proposed that for diagnosing WS II, two major criteria be fulfilled and dystopia canthorum be replaced with premature graying as one of the major criteria.
Several case reports have been published, some of them recently.[6],[7],[8] We report a young girl showing features of WS I with her pedigree chart showing variable penetrance of features and hence marked intrafamilial variation of expression of WS.
Case Report
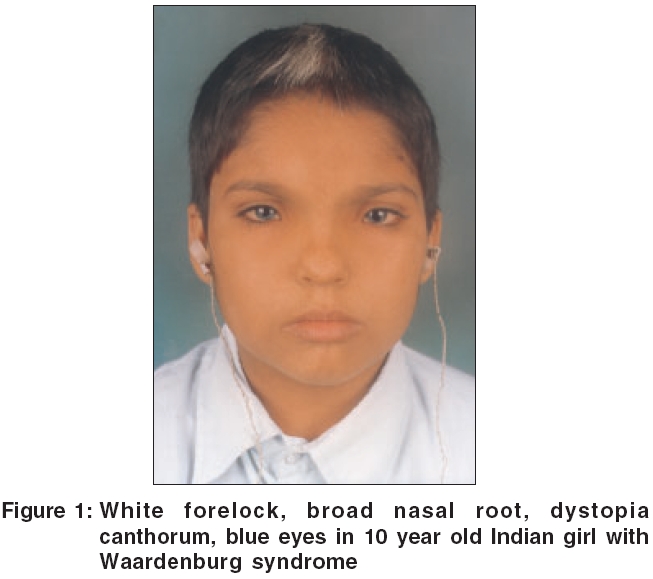
A 10 year old girl [Figure - 1] with white forelock and blue eyes was incidentally spotted at a boarding school for deaf and dumb when all the inmates were being examined during an outbreak of scabies. Blue color of her iridis was striking as compared to brown to black color normally seen in Indians. She had centrally placed white forelock in frontal area without any associated depigmentation of scalp or elsewhere on body. Her nasal root was broad with wide intercanthal distance. Girl had congenital hearing impairment with 110 dB loss in right ear and 105 dB loss in left ear. She was an intelligent, smart girl, well versed with sign language and lip reading and was doing well in her studies. On ophthalmological examination, palpebral fissure was smaller with lateral displacement of canthi and lacrimal punctae. Medially, sclera was visible to a lesser extent. She had hypoplastic blue iridis bilaterally. Pupils were mid dilated with sluggish reaction. Right fundus showed mottling while left fundus was albinotic. Inner canthal (a), interpupillary (b) and outercanthal distances (c) were measured using a rigid ruler held against face to calculate Waardenburg index (WI = X + Y + a/b) where X = (2a - 0.2119c - 3.909)/c and Y = (2a - 0.2479b - 3.909)/b. In our case, a = 40 mm, b = 54 mm, c = 83 mm and thus WI index was 2.6. We examined one maternal cousin of hers (V 14) who had hypoplastic blue iridis, canities, medial eyebrow flare with dystopia canthorum (a = 41 mm, b = 57 mm, c = 83 mm, WI = 2.57). Pedigree of index case was traced by interviewing her father and maternal uncle and we have used modified geneology symbols to depict presence of various features [Figure - 2]. Dystopia canthorum, medial eyebrow flare, broad nasal root and hypoplastic alae nasi could not be depicted as it was not possible to trace these features by history alone.
Discussion
Since the first description of this syndrome, there have been several speculations regarding genesis of this auditory-pigmentary complex characterized by physical absence of melanocytes affecting skin, hair, eyes or stria vascularis of cochlea.[1],[2] As all melanocytes except the retinal ones have their origin from the embryonic neural crest, Read and Newton[2] suggested that WS I, III and IV are neurocristopathies with additional involvement of frontal bone, limb muscles and enteric ganglia respectively. These tissues are also derived from neural crest. Alternately, absence of melanocytes can be melanocyte specific where melanoblasts may fail to migrate or differentiate terminally or survive in their final location. Some WS II may be melanocyte specific.[2] WS has been described as part of first arch syndrome as well.[9]
Dystopia canthorum is the most penetrant feature of WS and is found in 41.2 to 99% of the reported cases.[1] It presents with appearance of blepharophimosis and there is increased inner canthal distance with lacrimal punctae lying opposite cornea.[1],[2] Arias and Mota[10] developed a Waardenburg index (WI) as a reliable measure of dystopia canthorum. Earlier a WI> 2.07 was considered dystopic but now WI> 1.95 is considered more practical.[2]
Hearing loss is not a universal feature of WS but penetrance of sensorineural hearing loss has been observed to be 69% in WS I and 87% in WS II after excluding probands ascertained through their hearing loss.[3],[11]
Cutaneous pigmentary defects occur in 8.3 to 50% of patients.[1] Piebaldism like achromic spots may occur on face, trunk or limbs or even hyperpigmented macules may be seen.[1],[2] Hair changes may be in form of a white forelock, that usually involves the forehead but may occur elsewhere. Premature graying of scalp hair, eye brows, cilia or body hair has been reported to occur in 7% cases.[12]
Partial or complete heterochromia iridis occurs in 21 to 28% of patients with WS, hypoplastic blue iridis occur in 14.9 to 42% of cases.[1] Fundus may be albinotic or may show pigment mottling in the periphery.
Mutations in PAX3 gene on chromosome 2q37 have been observed in WS I and WS III. What impact a particular mutation has on the phenotype is not clear and symptoms vary even within families, hinting at interplay of some modifier genes.[1],[2] Read and Newton[2] have proposed that probably frontal bone development is most sensitive to PAX3 dosage, melanocytes to a lesser extent and limb buds least of all. It seems plausible keeping in view that frontal bone development defects occur in virtually all loss of function mutations in PAX3, while pigmentary defects and degree of hearing loss vary in affected individuals and limb defects of WS III occur in homozygotes mostly.
MITF gene is human analogue of the mouse mi (microphthalmia) gene and has been mapped to 3p12-p 14.1.[13] MITF has been proposed as a candidate gene in WS II.
Homozygous mutations in EDN3 or EDNRB occasionally show WS IV phenotype but heterozygotes are usually unaffected or have isolated Hirschsprung disease.[2]
All the insight into genetics of WS has not unraveled so far what final outcome is to be expected in an individual showing a mutation of candidate genes thus complicating the issue of genetic counselling and limiting the scope for prenatal diagnosis. All features of WSI and II except sensorineural hearing loss are essentially benign and cosmetic in nature and do not necessitate active intervention but deafness, if severe, can be a major handicap. An early diagnosis with social and vocational training and rehabilitation depending upon the extent of involvement remain the only recluse for the hearing impaired individuals.
| 1. |
Dourmishev AL, Dourmishev LA, Schwartz RA, Janniger CK. Waardenburg syndrome. Int J Dermatol 1999;38:656-63.
[Google Scholar]
|
| 2. |
Read AP, Newton VE. Waardenburg syndrome. J Med Genet 1997;34:656-65.
[Google Scholar]
|
| 3. |
Nayak CS, Isaacson G. Worldwide distribution of Waardenburg syndrome. Ann Otol Rhinol Laryngol 2003;112:817-20.
[Google Scholar]
|
| 4. |
Farrer LA, Grundfast KM, Amos J, Arnos KS, Asher JH Jr, Beighton P, et al . Waardenburg syndrome (WS) type 1 is caused by defects at multiple loci, one of which is near ALPP on chromosome 2: First report of the WS Consortium. Am J Hum Genet 1992;50:902-13.
[Google Scholar]
|
| 5. |
Liu XZ, Newton VE, Read AP. Waardenburg syndrome type 2: Phenotypic findings and diagnostic criteria. Am J Med Genet 1995;55:95-100.
[Google Scholar]
|
| 6. |
Kharya AV, Agarwal SN. Waardenburg's syndrome. Indian J Dermatol Venereol Leprol 2003;69:24-5.
[Google Scholar]
|
| 7. |
Krishtul A, Galadari I. Waardenburg syndrome: Case report. Int J Dermatol 2003;42:651-2.
[Google Scholar]
|
| 8. |
Denli YG, Yucel A, Gunasti S, Homan S, Durdu M, Suleymanova D. A family with Waardenburg syndrome. J Dermatol 2004;31:434-6.
[Google Scholar]
|
| 9. |
Campbell B, Campbell NR, Swift S. Waardenburg's syndrome. Arch Dermatol 1962;86:718-24.
[Google Scholar]
|
| 10. |
Arias S, Mota M. Apparent non-penetrance for dystopia in Waardenburg syndrome type I, with some hints in the diagnosis of dystopia canthorum. J Genet Hum 1978;26:103-31.
[Google Scholar]
|
| 11. |
Newton V. Hearing loss and Waardenburg syndrome: Implications for genetic counselling. J Laryngol Otol 1990;104:97-103.
[Google Scholar]
|
| 12. |
Goldberg MF. Waardenberg's syndrome with fundus and other anomalies. Arch Ophthalmol 1966;76:797-810.
[Google Scholar]
|
| 13. |
Hughes AE, Newton VE, Liu XZ, Read AP. A gene for Waardenburg syndrome type 2 maps to human chromosome 3p12-p14.1. Nat Genet 1994;7:509-12.
[Google Scholar]
|
Fulltext Views
6,695
PDF downloads
3,221





![[Table - 1]](#tbl_ijdvl_2006_72_4_326_26718_1.jpg){kind=link}
![[Figure - 1]](#fig_ijdvl_2006_72_4_326_26718_2.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2006_72_4_326_26718_3.jpg){kind=link}