Translate this page into:
Werner's syndrome
Correspondence Address:
Iffat Hassan
House No 35, Mominabad, Hyderpora, Srinagar, Jammu and Kashmir
India
| How to cite this article: Keen A, Hassan I. Werner's syndrome. Indian J Dermatol Venereol Leprol 2012;78:380-381 |
Sir,
Werner′s syndrome (pangeria or progeria adultorum) is a hereditary systemic disease in which the aging process is accelerated, starting after puberty. The clinical syndrome has an autosomal recessive mode of inheritance. Clinical characteristics include short stature, senile appearance, cataract, early menopause, premature arteriosclerosis, skin changes (scleroderma-like features, premature canities, baldness, and skin ulceration), associated with an increased risk of malignancy. [1] We present a case of Werner′s syndrome in view of its clinical rarity.
A 30-year-old unmarried female patient presented with baldness and premature graying of her hair since her mid-twenties. The patient reported that she had been short in stature since her childhood. She reported binding down and atrophy of skin over her hands, feet, and face. She also gave a history of recurrent non-healing ulcers over her joints. She had undergone bilateral cataract surgery at the age of 25 years and had amenorrhea since last one year.
Family history indicated first-degree consanguinity of her parents, but there were no symptoms in any of her siblings. General physical examination revealed short stature with an aged, bird-like face, and a beak-like nose [Figure - 1]. Her scalp hair was scanty and graying [Figure - 2].
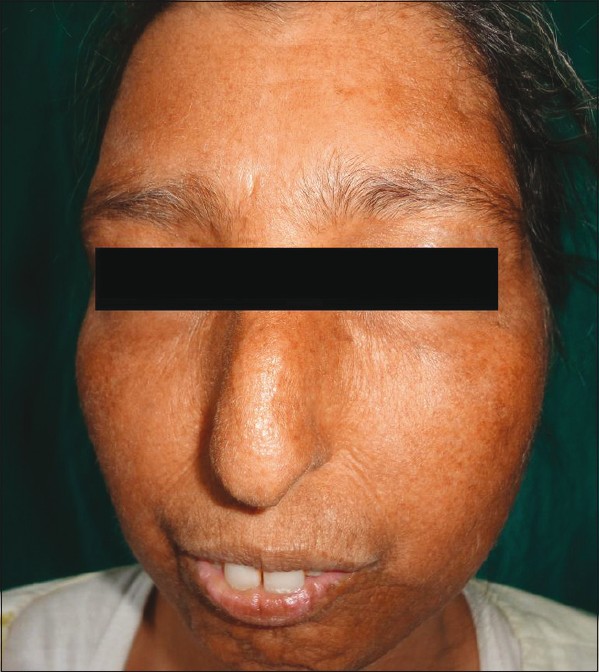 |
| Figure 1: Senile look with beak - like nose |
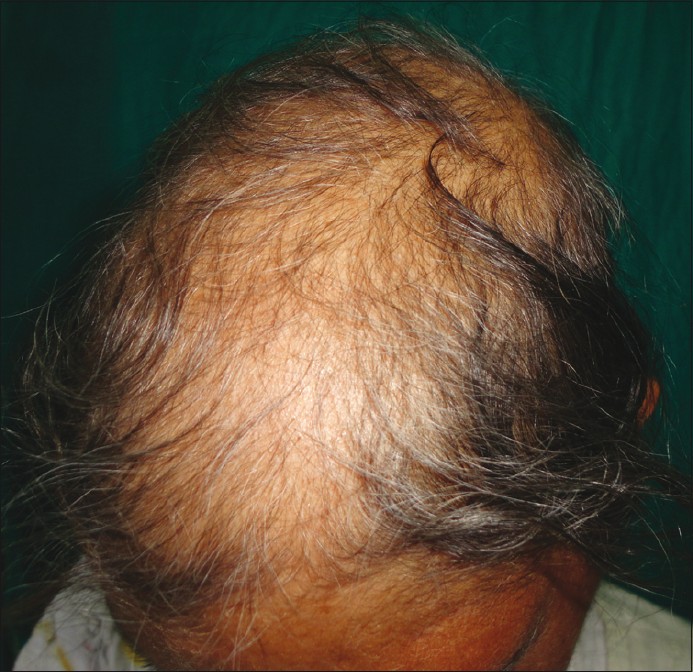 |
| Figure 2: Scanty and grey scalp hair |
The skin of the extremities was atrophic and sclerotic. There were keratoses of both feet and a single ill-defined crusted ulcerated plaque present over her right elbow region. Ophthalmological examination revealed aphakia in both eyes, with a corneal leukoma in her left eye. Gynecological examination of the patient was normal.
Routine laboratory and hematological investigations including a complete hemogram, liver function tests, renal function tests, fasting blood glucose, and fasting lipid profile were all normal. The Antinuclear Antibody (ANA) profile of the patient was negative. Radiographs of the limbs showed a generalized osteoporosis. Cardiovascular assessment including blood pressure monitoring, Electrocardiography (ECG), and echocardiography was done, the results of which were normal. Ultrasound of the abdomen and pelvis revealed a hypoplastic uterus. Serum Luteinizing Hormone (LH) and Follicle-Stimulating Hormone (FSH) levels were raised, while prolactin, 5-Dehydroepiandrosterone (DHEAS), and testosterone levels were normal. Genetic analysis was not carried out because of lack of facilities for the same. Skin biopsy was also not done as the patient was reluctant for the same. Our patient had all classical features of Werner′s syndrome. The patient was put on oral vitamin C and advised regular follow-up.
Werner′s syndrome was initially described by Werner′s in 1904, when he reported four cases of brothers and sisters with symptoms and signs including juvenile cataract, pachyderma-like alteration of the extremities, short stature, premature aging of the face, juvenile grey hair, and genital hypoplasia. The syndrome is due to an autosomal recessive gene with a gene frequency of 1-5 per 1000 population. [2] Causal mutation has been identified in the Rec Q type DNA helicase gene (REC QL 2, WRN gene), which has been observed to play a significant role in DNA replication, repair, and telomere maintenance. [3] The most important complications associated with Werner′s syndrome are arteriosclerosis and malignancies. The most common malignancy seen in patients of Werner′s syndrome is fibrosarcoma, which occurs in 10% of the patients. [4] Other malignancies reported are osteosarcoma, leukemia, malignant soft tissue tumor, rhabdomyosarcoma, leiomyosarcoma, thyroid cancers, squamous cell carcinoma, basal cell carcinoma, malignant melanoma, breast carcinoma, and gastric carcinoma. Therefore, it is imperative to recognize Werner′s syndrome at an early stage so as to facilitate the identification of subsequent malignant tumors. [5] Death usually occurs in the fourth to sixth decade, due to myocardial infarction or malignancy.
In the differential diagnosis of Werner′s syndrome, progeria, acrogeria, and Rothmund-Thomson syndrome are included. Progeria is a rare combination of dwarfism, premature aging, sclerodermatous changes, and alopecia. Acrogeria is a progeroid syndrome of the skin without the involvement of internal organs. Rothmund-Thomson syndrome is a rare autosomal dominant disorder characterized by photosensitivity, poikilodermatous skin changes, juvenile cataracts, skeletal dysplasia, and a predisposition to osteosarcoma, and skin cancer.
There is no specific treatment for this condition and only symptomatic measures are available. The skin ulcers require an aggressive management. In some cases, amputation may be needed. Recently, bosentan has been used in the management of leg ulcers in Werner′s syndrome. [6] Surgery should be undertaken for ocular cataracts. Diabetes mellitus and hyperlipidemia, if present, should be adequately addressed. Results of various animal studies suggest that vitamin C supplementation could be beneficial for patients with Werner′s syndrome.
| 1. |
Epstein CJ, Martin GM, Schultz AL, Motulsky AG. Werner's syndrome: A review of its symptomatology, natural history, pathologic features, genetics and relationship to the natural aging process. Medicine (Baltimore) 1996;45:177-221.
[Google Scholar]
|
| 2. |
Yu CE, Oshima J, Wijsman EM, Nakura J, Miki T, Piussan C, et al. Mutations in the concensus helicase domains of the Werner's syndrome gene. Werner's Syndrome Collaborative Group. Am J Hum Genet 1997;60:330-41.
[Google Scholar]
|
| 3. |
Tannhauser SJ. Werner's syndrome (progeria of the adult) and Rothmund's syndrome: Two types of closely related heredofamilial atrophic dermatoses with juvenile cataracts and endocrine features; a critical study with five new cases. Ann Intern Med 1945;23:559-626.
[Google Scholar]
|
| 4. |
Fujiwara Y, Higashikawa T, Tatsumi M. A retarded rate of DNA replication and normal level of DNA repair in Werner's syndrome fibroblasts in culture. J Cell Physiol 1997;92:365-74.
[Google Scholar]
|
| 5. |
Hrabko RP, Milgrom H, Schwartz RA. Werner's syndrome with associated malignant neoplasms. Arch Dermatol 1982;118:106-8.
[Google Scholar]
|
| 6. |
Noda S, Asano Y, Masuda S, Miyagawa T, Sugita M, Yamamoto M, et al. Bosentan: A novel therapy for leg ulcers in Werner's syndrome. J Am Acad Dermatol 2011;65:54-5.
[Google Scholar]
|
Fulltext Views
6,706
PDF downloads
3,583





![[Figure - 1]](#fig_ijdvl_2012_78_3_380_95469_f1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2012_78_3_380_95469_f2.jpg){kind=link}