Translate this page into:
Widespread hyperpigmented and hyperkeratotic plaques following lines of Blaschko
2 Department of Pathology, Cardiovascular Training Education, Van, Turkey
Correspondence Address:
Serap Gunes Bilgili
Department of Dermatology, Yuzuncu Yil University, 65100 Van
Turkey
| How to cite this article: Bilgili SG, Karadag AS, Calka O, Bulut G, Cecen I. Widespread hyperpigmented and hyperkeratotic plaques following lines of Blaschko. Indian J Dermatol Venereol Leprol 2012;78:403-405 |
A 3-year-old girl was admitted to our outpatient clinic with complaints of brown cutaneous verrucous lesions. Three months after her birth, lesions had first appeared around her navel and then gradually spread over to the trunk. Her parents described occasional blister formation that were located on the trunk and limbs. Her parents were nonconsanguineous and had no family history of similar disorders.
Dermatological examination revealed linearly distributed papules and hyperkeratotic plaques along Blaschko′s lines with areas of scaling and crusting over lower extremities, inguinal region, buttocks, abdomen and scapular region [Figure - 1] and [Figure - 2]. There were not any lesions in the skin creases. Hair, nail and teeth problems, abnormality in gait, digital contractures were not seen in the case. Neurological examination was normal.
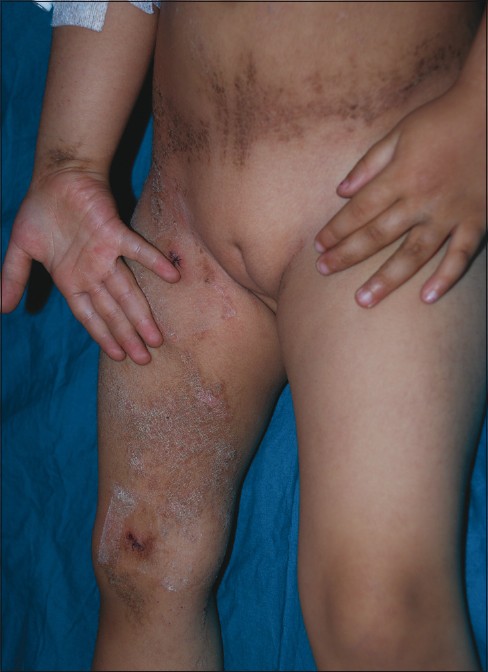 |
| Figure 1: Hyperkeratotic papules and plaques with areas of scaling and crusts over lower limb, inguinal region and abdomen |
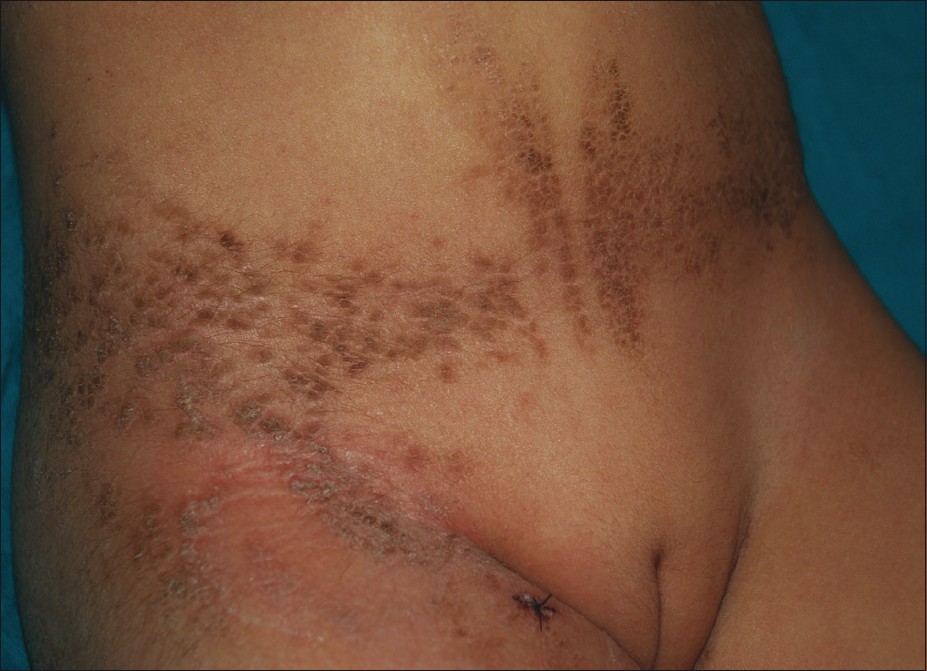 |
| Figure 2: Hyperkeratotic papules and plaques on the abdomen |
Histopathological examination of a biopsy taken from the lesion showed compact hyperkeratosis, papillomatosis, degeneration and prominent keratohyalin granules in the granular layer. There were perinuclear clear spaces in variable sizes in stratum granulosum and stratum spinosum with faint keratohyalin granules at their peripheries. Focal vacuolar degeneration in the basal layer, perivascular lymphocytic infiltration and sparse melanophages were detected in the upper dermis [Figure - 3].
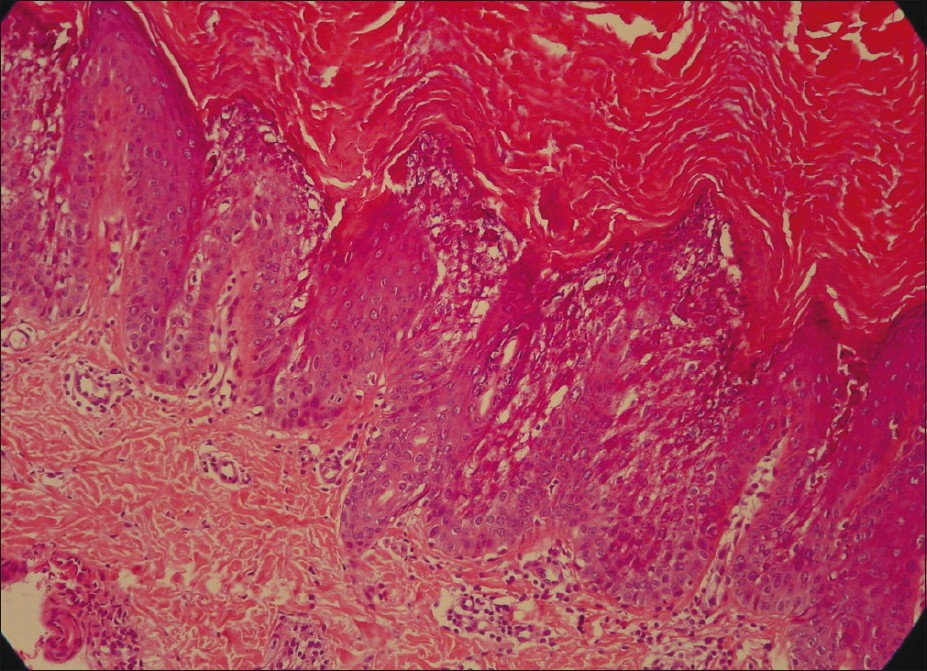 |
| Figure 3: Histopathology showing vacuolization of the upper and mid spinous layer, hyperkeratosis with large keratohyaline granules in the vacuoled expanded granular cell layer (H and E, ×200) |
What is Your Diagnosis?
Answer: Epidermolytic hyperkeratosis (EHK)
Discussion
Epidermolytic hyperkeratosis (bullous congenital ichthyosiform erythroderma) is a rare autosomal dominant disorder of keratinization with an estimated prevalence of 1:200,000. [1],[2] Mutations in the genes that encode keratin-1 and keratin-10 have been associated with this disorder(OMIM #113800). [1] Mutations are mapped on the keratin gene clusters within chromosomes 12q and 17q. The frequency of sporadic cases due to spontaneous mutations is as high as 50%. [3] Patients characteristically develop generalised erythema, scaling and blistering that occur especially at birth or in the early childhood. Gradually hyperkeratotic plaques develop and become more prominent around large joints. They exhibit intraepidermal blisters and erosions at sites of even mild trauma. EHK shows a variable clinical course in terms of cutaneous manifestations such as hyperkeratosis, blistering, the presence of erythroderma, and the involvement of palms and soles. [4] Disease generally has a more severe course in early childhood.
Mosaic EHK is due to a postzygotic, spontaneous mutation. Hyperkeratosis is distributed in streaks along Blaschko′s lines. It is not inherited unless postzygotic mutation occurs in the germ line. It may be difficult to distinguish mosaic EHK from epidermal nevus clinically without characteristic histopathological features which are the same as those of the generalized EHK. [5] EHK is divided into two groups, based on presence or absence of severe palm/sole involvement (PS type or NPS type). Each group consists of three sub-groups totalling six distinct clinical phenotypes. [6] Our patient had no palmar/plantar hyperkeratosis. Her past medical history was negative for erythroderma. Diagnosis of NPS-type EHK was made according to the clinical and histopathological findings.
Histopathology of epidermolytic hyperkeratosis is characterized by hyperkeratosis, hypergranulosis, and epidermolysis. Histopathology demonstrates perinuclear vacuolization with indistinct peripheral boundaries. Keratohyalin granules are basophilic and clumped. On histopathological assessment keratinocytes with indistinct cell boundaries exhibiting variously sized clear spaces around their nuclei are noted within the upper spinous and granular layers. [5] Mosaic EHK is focal epidermolytic hyperkeratosis with epidermolytic hyperkeratosis horizontally alternating with areas without epidermolytic hyperkeratosis. [5] In our patient, histopathological findings were consistent with mosaic EHK.
Ichthyosis bullosa of Siemens and ichthyosis hystrix of Curth and Macklin should be considered in the differential diagnosis for EHK. Ichthyosis bullosa of Siemens is different than EHK, as the epidermal fragility is very superficial and hyperkeratosis and vacuolization are confined to the granular layer in the latter. Ichthyosis hystrix of Curth and Macklin presents with extensive and very severe, spiky or verrucous hyperkeratosis without blisters. Frequently hyperkeratosis is more prominent over the extensor surfaces of the arms and legs. [6] ILVEN (Inflammatory linear verrucous epidermal nevus), Darier disease, hypertrophic lichen planus, incontinentia pigmenti should also be taken into account in the differential diagnosis of EHK as in our case.
Various treatment options exist for managing EHK including of keratolytics, antiseptic washes, topical calcipotriol, alpha hydroxy acids and oral retinoids. [6] Unfortunately current treatment modalities are not sufficiently effective. In our patient topical treatment with 10% salicylic acid along with a moisturizer cream was commenced but discontinued after a week because of the lack of improvement with treatment. We initiated 0.1% tretinoin cream mixed with the same amount of petrolatum by weight. Mixture was applied every night. Due to an inadequate response to treatment we planned and recommended to start on oral acitretin treatment but parents of the patient did not consent to treatment.
EHK is a rare disease. It is recognized by its distinctive histopathological features. It is very important to inform patients and their relatives about the course and prognosis of the disease in detail.
| 1. |
Chassaing N, Kanitakis J, Sportich S, Cordier-Alex MP, Titeux M, Calvas P, et al. Generalized epidermolytic hyperkeratosis in two unrelated children from parents with localized linear form, and prenatal diagnosis. J Invest Dermatol 2006;126:2715-7.
[Google Scholar]
|
| 2. |
Pulkkinen L, Christiano AM, Knowlton RG, Uitto J. Epidermolytic hyperkeratosis (bullous congenital ichthyosiform erythroderma). Genetic linkage to chromosome 12q in the region of the type II keratin gene cluster. J Clin Invest 1993;91:357-61.
[Google Scholar]
|
| 3. |
Betlloch I, Lucas Costa A, Mataix J, Pérez-Crespo M, Ballester I. Bullous congenital ichthyosiform erythroderma: A sporadic case produced by a new KRT10 gene mutation. Pediatr Dermatol 2009;26:489-91.
[Google Scholar]
|
| 4. |
Math A, Frank J, Handisurya A, Poblete-Gutiérrez P, Slupetzky K, Födinger D, et al. Identification of a de novo keratin 1 mutation in epidermolytic hyperkeratosis with palmoplantar involvement. Eur J Dermatol 2006;16:507-10.
[Google Scholar]
|
| 5. |
Ross R, DiGiovanna JJ, Capaldi L, Argenyi Z, Fleckman P, Robinson-Bostom L. Histopathologic characterization of epidermolytic hyperkeratosis: A systematic review of histology from the National Registry for Ichthyosis and Related Skin Disorders. J Am Acad Dermatol 2008;59:86-90.
[Google Scholar]
|
| 6. |
Nayak S, Behera SK, Acharjya B, Sahu A, Mishra D. Epidermolytic hyperkeratosis with rickets. Indian J Dermatol Venereol Leprol 2006;72:139-42.
[Google Scholar]
|
Fulltext Views
7,163
PDF downloads
3,462





![[Figure - 1]](#fig_ijdvl_2012_78_3_403_95475_f1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2012_78_3_403_95475_f2.jpg){kind=link}
![[Figure - 3]](#fig_ijdvl_2012_78_3_403_95475_f3.jpg){kind=link}