Translate this page into:
Bloom syndrome sans characteristic facial features in a Mestizo patient- a diagnostic challenge
2 Department of Genetics, Hospital Universitario “Dr. José Eleuterio González” UANL, Monterrey, Nuevo León, Mexico
Correspondence Address:
Luis Daniel Campos-Acevedo
Hospital Universitario “Dr. José Eleuterio González” UANL, Avenida Francisco I. Madero y Gonzalitos s/n Colonia Mitras Centro, C.P. 64460, Monterrey, Nuevo León
Mexico
| How to cite this article: Chavez-Alvarez S, Villarreal-Martinez A, Velasco-Campos MR, Moreno-Vega I, Martinez-de-Villarreal LE, Herz-Ruelas M, Ocampo-Candiani J, Campos-Acevedo LD. Bloom syndrome sans characteristic facial features in a Mestizo patient- a diagnostic challenge. Indian J Dermatol Venereol Leprol 2019;85:130 |
Sir,
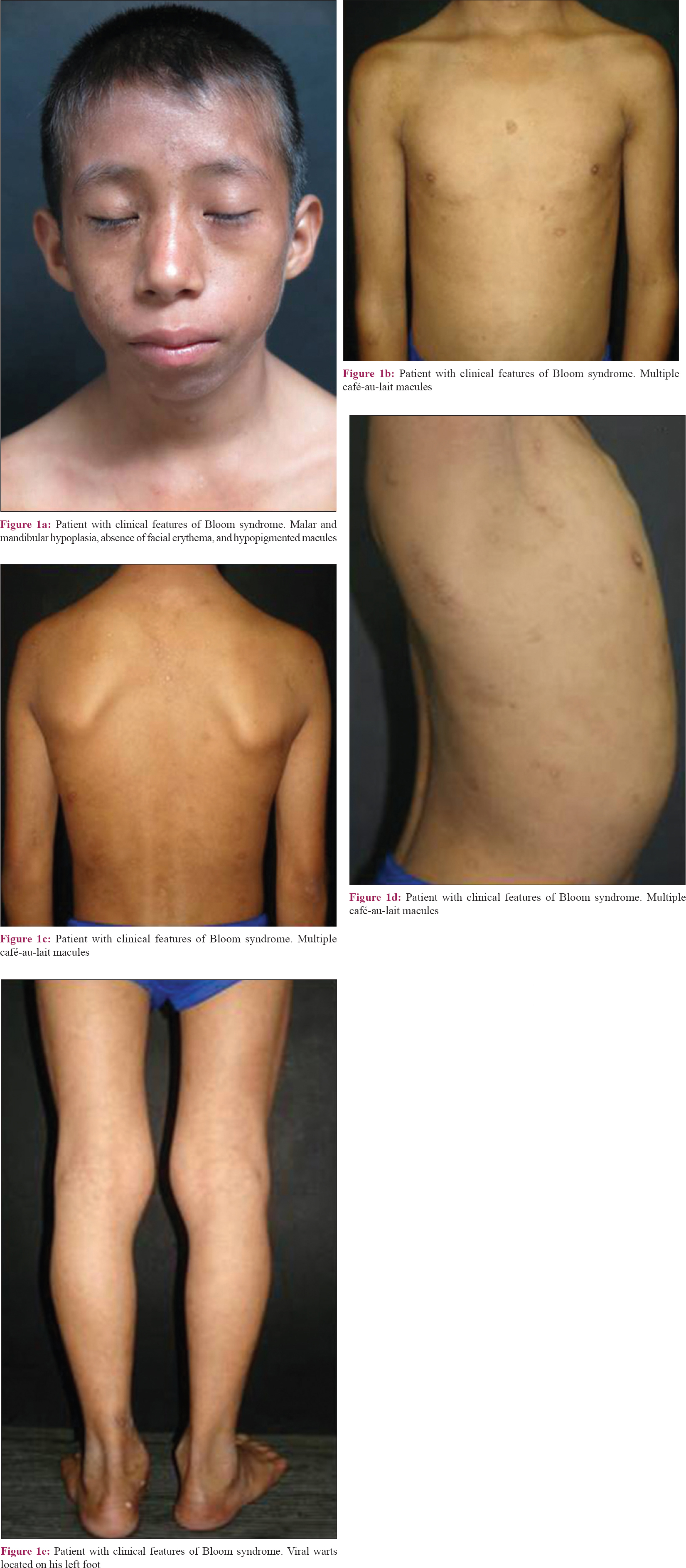
Bloom syndrome is a rare dermatosis, clinically diagnosed by characteristic cutaneous features, many of them affecting the face. The expected phenotype is a small for age patient with micrognatia, dolichocephaly, malar erythema, photosensitivity, multiple café-au-lait macules, and immunodeficiency. Here we present a 12-year-old Mestizo patient with clinical features of Bloom syndrome devoid of malar erythema and photosensitivity. This case illustrates that Bloom syndrome should be suspected even in the absence of hallmark facial erythema, especially in the Latin and Asian populations.
Bloom syndrome is an autosomal recessive genodermatosis presenting with characteristic cutaneous manifestations like photosensitivity, facial telangiectasias and erythema, and café-au-lait macules. Individuals with Bloom syndrome are short statured, immunodeficent and are more prone to develop malignancies.
A 12-year-old Mexican male patient, born out of non-consanguineous parentage, presented to our Genetics Department with complaints of delayed growth. He had a pre-term birth at 35 weeks of gestation from an unplanned normal pregnancy, with a weight of 1.9 kg (P 90) and a crown-heel length of 37 cm (P < 3). He was hospitalized with pneumonia at the age of 3 months and had suffered from recurrent respiratory tract infections and intermittent noninflammatory diarrhoea. No evidence of photosensitivity was obtained. Family history was normal with non-affected siblings. Currently, the patient measures 120 cm and weighs 18.5 kg; both parameters falling below the 3rd percentile on growth charts. Physical examination revealed dolichocephaly, mandibular hypoplasia, and a high-pitched voice. Dermatologic findings included skin phototype IV without facial erythema or telangiectasias, several café-au-lait macules on his trunk and viral warts on his left foot [Figure - 1]. Blood biochemistry was unremarkable. An X-ray of both hands revealed a bone age of 10 years.
 |
| Figure 1 |
Despite the lack of characteristic photosensitivity and facial telangiectasias/erythema (not visible even on dermoscopic examination) Bloom syndrome was suspected because of the presence of other distinct features like short stature, sharp voice, microcephaly, delayed bone age, chronic intermittent diarrhoea, malar hypoplasia, triangular face shape, café-au-lait macules, and a history of recurrent upper respiratory tract infections. Dubowitz syndrome was considered as a differential because it also presents with short stature, microcephaly, long face, recurrent infections, chronic diarrhoea, and delayed bone age, however, café-au-lait macules favoured our diagnosis.
Homologous chromatid exchange and a karyotype confirmed the diagnosis. A micronucleus test was performed for the chromatid study. Micronuclei (mn) originate from chromosome fragments or whole chromosomes that are not included in the main daughter nuclei during nuclear division as a result of chromosome breakage caused by unrepaired or mis-repaired DNA lesions or chromosome mal-segregation because of mitotic dysfunction. Chromosome breakage may be precipitated by oxidative stress, genetic defects in cell cycle checkpoint, and/or DNA repair genes, or deficiencies in nutrients required as cofactors in DNA metabolism and chromosome segregation machinery. Micronuclei provoke chromosome instability. The Karyotype of our case was 46, X, chrb (2) (q21)/46, XY, chrb (5) (q31)/46, XY, chrb (q22)/46, XY. Sister chromatid exchange was carried out with a Mexican male control. The chromatid sister exchange and karyotype was performed based on clinical data. We could not perform the molecular test for BLM gene or IQ test due to lack of resources.
He was counselled about his condition and instructed to apply SPF 50 sunscreen cream and adopt other physical photoprotective measures. Annual medical check-ups have been suggested [Figure - 2].
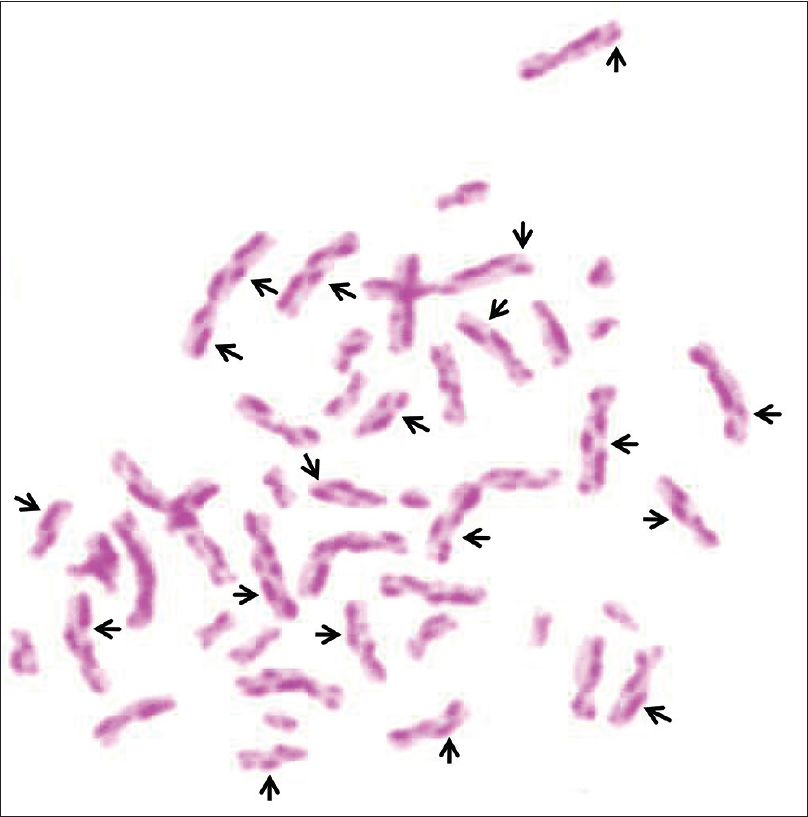 |
| Figure 2: Chromatid sister exchange performed in peripheral blood |
Telangiectatic facial erythema and short stature were recognized as the classical clinical features in 1965 in individuals with Bloom syndrome.[1] This genodermatosis results from chromosomal instability in the BLM gene and an increase in the chromatid sister exchange.[2] The increase in chromatid sister exchange is characteristic of Bloom syndrome.
Patients have severe growth retardation, dolichocephaly, and malar and mandibular hypoplasia with a prominent nose.[3] Cutaneous signs include photosensitivity, facial erythema with butterfly distribution, telangiectasias on sun-exposed skin, café-au-lait macules, hypopigmented macules, chronic cheilitis, and madarosis.[3] Other features include normal intelligence, a high-pitched voice, a highly arched palate, congenital heart disease, annular pancreas, primary hypogonadism, clinodactyly, and syndactyly.[4] Males are affected more than females (1.3:1).[5] Our patient presented with similar clinical features except photosensitivity and facial malar erythema.
The differential diagnoses include Rothmund–Thomson syndrome, Cockayne syndrome, hereditary hemorrhagic telangiectasia, xeroderma pigmentosum, and fucosidosis type III.[3],[5] All these differentials have been ruled out as chromosomal analysis of our case confirmed Bloom's syndrome.
Immunodeficiency predisposes these patients to infections particularly affecting the ears and lungs.[1] Malignancy is frequent, usually presenting with uncommon tumors such as osteosarcoma, medulloblastoma, and Wilms' tumor.[6],[7] These individuals may present with more than one cancer, and their life expectancy is about 30 years.[8]
Females and dark skin phototypes exhibit fewer cutaneous manifestations.[9] An absence of facial erythema/telangiectasias and photosensitivity has also been reported in Japanese patients[2], similar to our patient In Latin America, Bloom syndrome resulted from Jewish migration from Spain. This ethnic diversity may be responsible for the clinical variations,[10] in our case. There are only 300 registered cases of Bloom syndrome in the world, but only another Mestizo patient reported with evidence of photosensitivity. It is useful to keep in mind that this genetic heterogeneity may alter the typical presentation of Bloom syndrome, and this phenotype could be due to the variable gene expression.
This case has been reported to generate awareness about the atypical presentation of this uncommon disorder.
The authors certify that they have obtained all appropriate patient consent forms. In the form, the legal guardian has given his consent for images and other clinical information to be reported in the journal. The guardian understands that names and initials will not be published and due efforts will be made to conceal patient identity, but anonymity cannot be guaranteed.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient has given his consent for his images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
| 1. |
German J. Bloom's syndrome. Dermatologic Clinics 1995;13:7-18.
[Google Scholar]
|
| 2. |
Weksberg R, Smith C, Anson-Cartwright L, Maloney K. Bloom syndrome: A single complementation group defines patients of diverse ethnic origin. American Journal Of Human Genetics 1988;42:816-24.
[Google Scholar]
|
| 3. |
Sahn EE, Hussey RH, Christmann LM. A Case of Bloom Syndrome with Conjunctival Telangiectasia. Pediatric Dermalology Journal 1997;14:120-4.
[Google Scholar]
|
| 4. |
Sultan SJ, Sultan ST. Bloom syndrome in two siblings. Pediatric Dermatology 2010;27:174-7.
[Google Scholar]
|
| 5. |
Shah H, Sheth FJ, Pandit VS, Langanecha B. Bloom syndrome: report of two cases in siblings. International Journal of Dermatology 2013;52:990-2.
[Google Scholar]
|
| 6. |
Ben Salah G, Salem IH, Masmoudi A, Ben Rhouma B, Turki H, Fakhfakh F, et al. Chromosomal instability associated with a novel BLM frameshift mutation (c.1980-1982delAA) in two unrelated Tunisian families with Bloom syndrome. Journal of the European Academy of Dermatology and Venereology: JEADV. 2014;28:1318-23.
[Google Scholar]
|
| 7. |
Draznin M, Robles DT, Nguyen V, Berg D. An unusual case of Bloom syndrome presenting with basal cell carcinoma. Dermatologic surgery: Official publication for American Society for Dermatologic Surgery [et al]. 2009;35:131-4.
[Google Scholar]
|
| 8. |
German J, Sanz MM, Ciocci S, Ye TZ, Ellis NA. Syndrome-causing mutations of the BLM gene in persons in the Bloom's Syndrome Registry. Human Mutation 2007;28:743-53.
[Google Scholar]
|
| 9. |
Rivera H, Garcia-Cruz D, Vaca G, Moller M, Ramos-Zepeda R, Cantu JM. Bloom syndrome in a Mexican mestizo girl. Annales de Genetique 1986;29:39-41.
[Google Scholar]
|
| 10. |
Nuno-Arana I, Garcia-Garcia VA, Espejo-Plascencia I, Ramos-Zavala AL, Rivera H. An intracranial carcinoma in a Mexican woman with Bloom syndrome. Annales de Genetique 2000;43:55-7.
[Google Scholar]
|
Fulltext Views
4,767
PDF downloads
2,911





![[Figure - 1]](#fig_ijdvl_2019_85_1_130_232982_f1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2019_85_1_130_232982_f2.jpg){kind=link}