Translate this page into:
An uncommon presentation of Galli–Galli disease
2 Sakhiya Skin Clinic Pvt. Ltd., Surat, Gujarat, India
Correspondence Address:
Chirag Ashwin Desai
B21, 6th Floor, Krishnalaya Building, N S Mankikar Marg, Chunabhatti, Mumbai - 400 022, Maharashtra
India
| How to cite this article: Desai CA, Virmani N, Sakhiya J, Khopkar U. An uncommon presentation of Galli–Galli disease. Indian J Dermatol Venereol Leprol 2016;82:720-723 |
Sir,
Galli–Galli disease is a rare genodermatosis, also described as the acantholytic variant of Dowling–Degos disease. It is inherited in an autosomal dominant pattern and is caused by a mutation in the keratin 5 gene.[1],[2],[3]
A 55-year-old woman presented with multiple hyperpigmented lesions widely distributed on her body since early adolescence. These lesions were mostly asymptomatic with occasional itching that worsened in hot and humid conditions. On further inquiry, the patient acknowledged the presence of similar lesions in many family members of different generations, none of whom had sought medical advice. Analysis of the pedigree chart revealed that these lesions were more commonly seen among women, all of whom had a similar age of onset [Figure - 1]. On examination, there was mottled hyperpigmentation on the neck, chest, abdomen and extensor as well as flexor aspects of both the upper and lower extremities [Figure - 2] and [Figure - 3]. Moreover, there were multiple flat-topped hyperpigmented papular lesions. Some hyperpigmented macules were seen on the face. A provisional diagnosis of reticulate pigmentary disorders was considered, including Dowling–Degos disease and Galli–Galli disease.
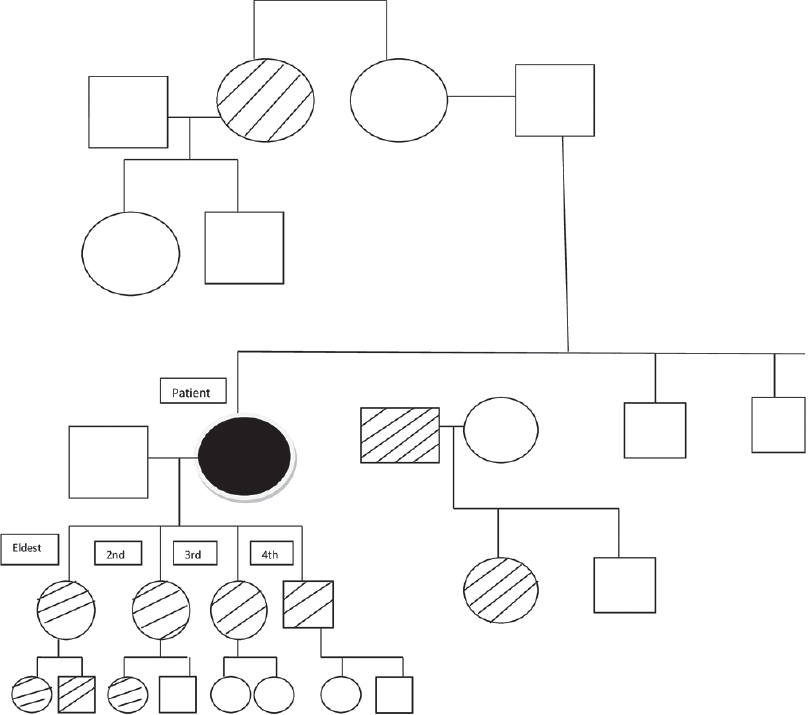 |
| Figure 1: Pedigree chart of the family |
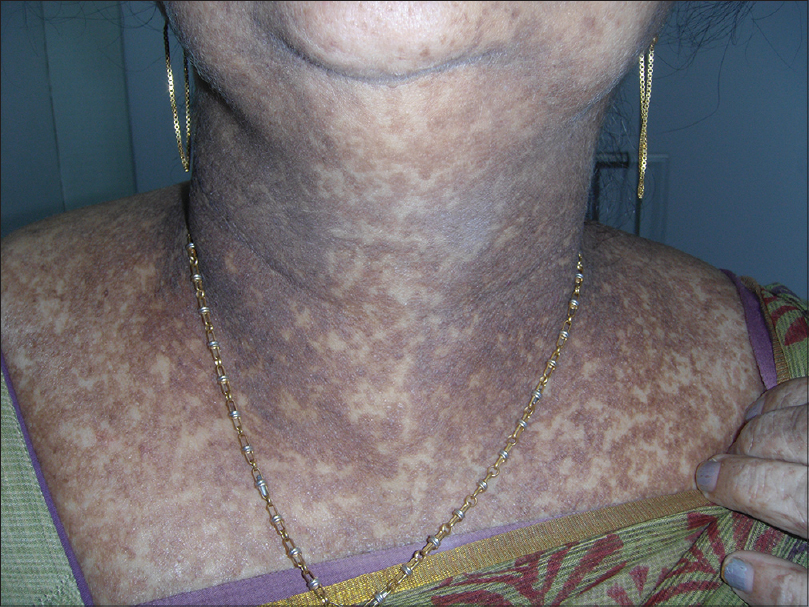 |
| Figure 2: Hyperpigmented macules distributed in a reticulate pattern on the neck |
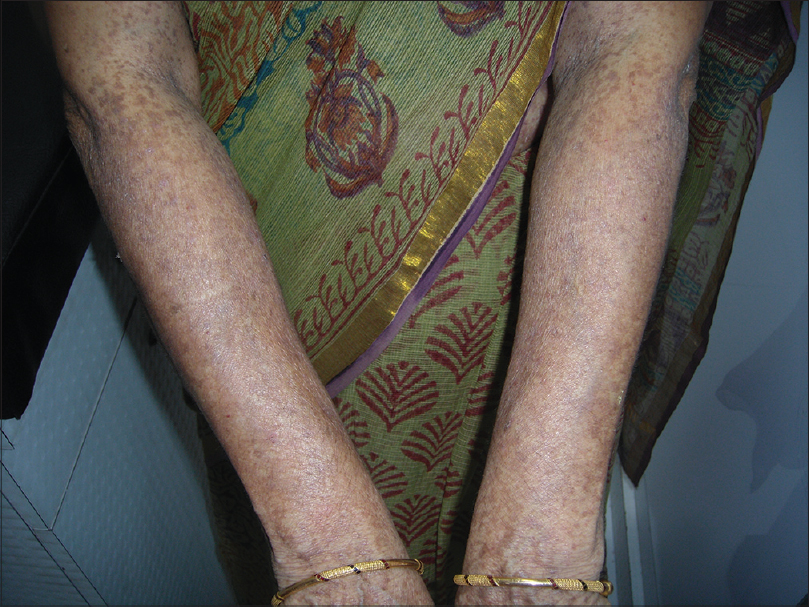 |
| Figure 3: Hyperpigmented macules on the dorsal aspect of the forearms |
A biopsy of one of the papular lesions revealed elongated rete ridges with increased melanization of basal layer. There was a broad zone of suprabasal acantholytic clefting of the epidermis with discrete overlying parakeratosis without dyskeratosis. A mild superficial perivascular lymphohistiocytic infiltrate was found in the dermis [Figure - 4] and [Figure - 5]. No “dilapidated brick wall” appearance, typical of Hailey–Hailey disease was seen. A repeat biopsy revealed that these findings were consistently present [Figure - 6]. We came to a final diagnosis of Galli–Galli disease in the patient.
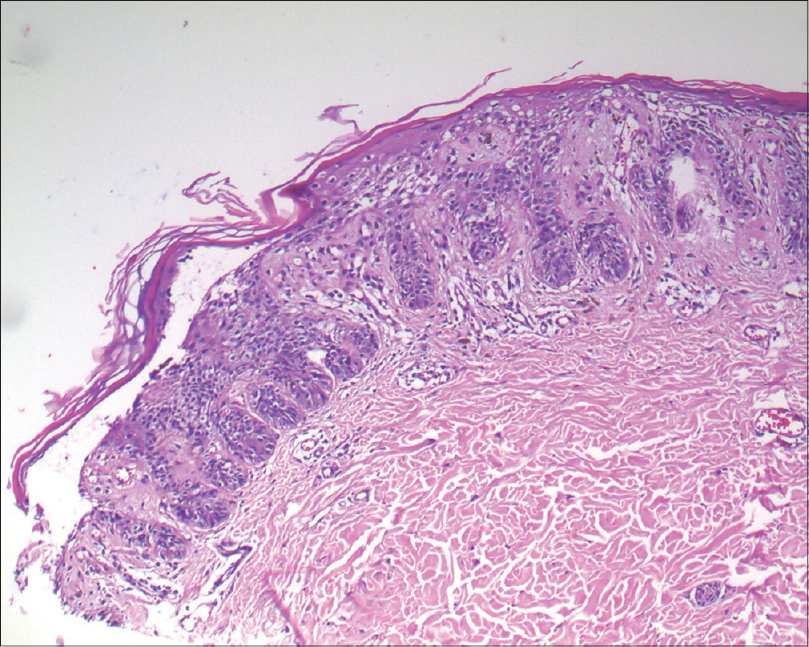 |
| Figure 4: Broad zone of suprabasal acantholytic split in the epidermis in the biopsy taken from the neck of the patient (H and E, ×100) |
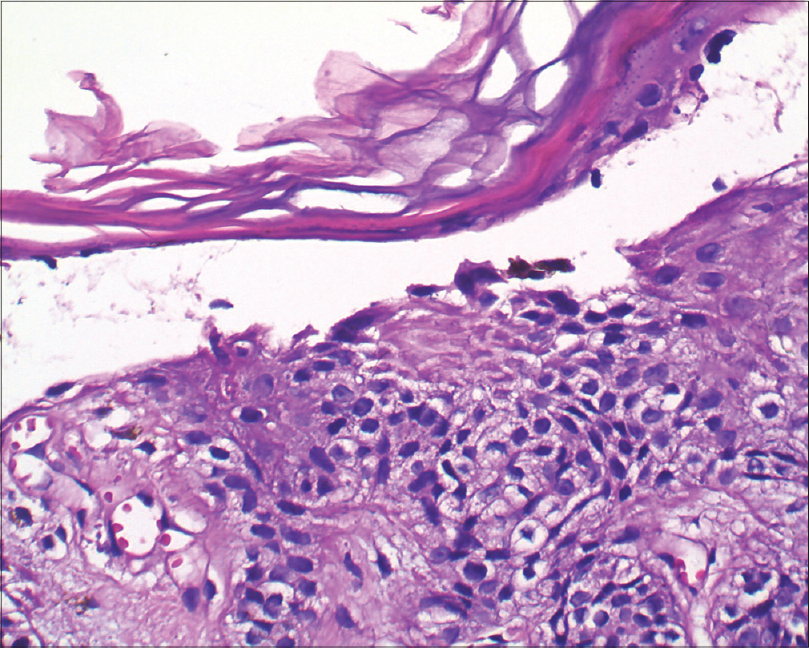 |
| Figure 5: Split with a few acantholytic cells (H and E, ×400) |
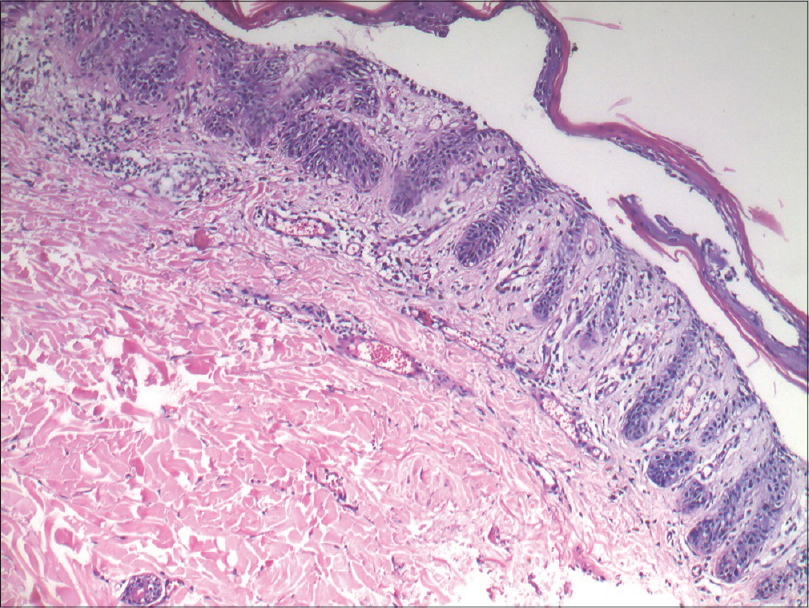 |
| Figure 6: Repeat biopsy from the same patient showing a similar acantholytic split (H and E, ×400) |
Following this, her 35-year-old niece also presented to us with asymptomatic, hyperpigmented macules localized to the face, neck and upper arms. These lesions had been unaltered in size and number since their onset in early adolescence [Figure - 7]. Histopathological examination of the lesions from the neck revealed 'antler-horn' like rete ridges with hypermelanization of the basal layer of the epidermis and multiple horn cysts, consistent with a diagnosis of Dowling–Degos disease [Figure - 8].
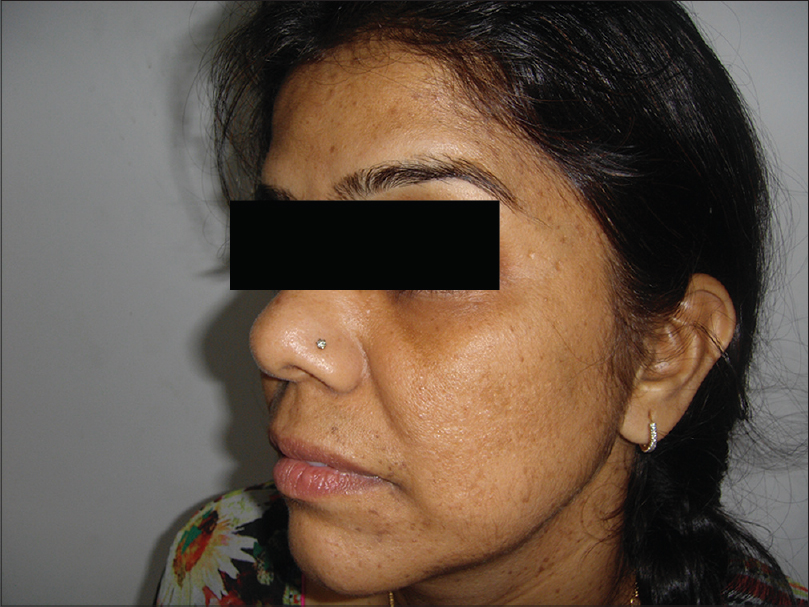 |
| Figure 7: Hyperpigmented macules on the face and neck of the patient's niece |
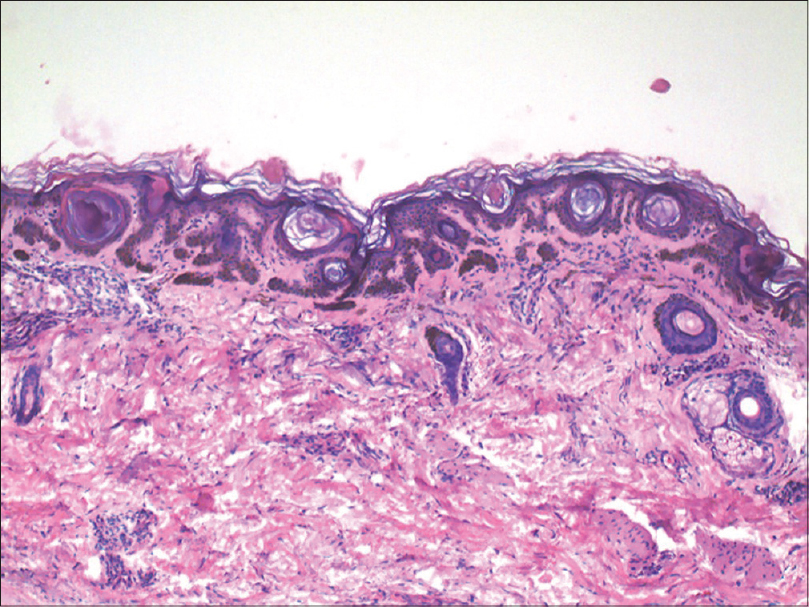 |
| Figure 8: Antler horn-like rete ridges with hypermelanization of epidermis with multiple horn cysts in biopsy from patient's niece (H and E, ×100) |
Galli-Galli disease, a histologic variant of Dowling–Degos disease, was first described by Bardach et al. in 1982, in two siblings. Galli–Galli disease and Dowling–Degos disease are clinically indistinguishable and the only differentiating element is finding areas of acantholysis, usually without dyskeratosis.[4] Although the inheritance pattern is autosomal dominant with variable penetrance, there have been reports of cases which lack a family history of this disorder.[2],[3] This disease predominantly appears between adolescence and middle age, though a few reports of onset in children with a family history of the disease have been documented.[5]
Mutations in the keratin 5 gene have been known to cause structural modifications in the intermediate filaments and have been linked with disease causation. However, these findings are not present in all patients diagnosed with the disease.[6]
The disease usually presents with hyperpigmented macules in the skin folds. Other findings include papules, scaly erythematous plaques, comedo-like lesions and pitted perioral scars. In our case, hyperpigmented macules and a few papules were present; however, the disease was not limited only to the skin folds, it was more generalized. A previously reported case showed only papular lesions in the skin folds and other sites without any macular pigmentation.[7]
Moreover, there are several previous reports showing overlap between the different reticulate pigmentary disorders such as reticulate acropigmentation of Kitamura, acropigmentation of Dohi, Haber syndrome, dyschromatosis universalis hereditaria, dyschromatosis symmetrica hereditaria and Dowling–Degos disease in the same patients.[8],[9] The overlap between Galli–Galli disease and Dowling–Degos disease in a patient is also reported: biopsy from the axillary lesion showed acantholysis while biopsies from the other sites did not.[8],[9] This indicates that both classic and acantholytic variants can coexist in the same patient or that the acantholytic variant is simply an incidental finding in typical Dowling–Degos disease. The phenomenon of Galli–Galli disease and Dowling–Degos disease coexisting in families has also been previously reported in an Indian study.[10] The finding of an acantholytic split in both biopsies from our first patient proved that the split was not an artifact and confirmed our diagnosis of Galli–Galli disease in the patient. The typical findings of Dowling–Degos disease in the patient's niece shows that these two variants can coexist in the same family. Treatment of this disease includes topical and systemic retinoids and phototherapy with only partial or no relief. We treated our patient with topical tretinoin.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has given her consent for her images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
| 1. |
García-Salces I, Hörndler C, Requena L. Galli-Galli disease presenting as lichenoid papules in the flexures. Actas Dermosifiliogr 2010;101:168-72.
[Google Scholar]
|
| 2. |
Ackerman AB, Brunhoeber P. Further references of Galli-Galli disease. J Am Acad Dermatol 2008;59:531-2.
[Google Scholar]
|
| 3. |
Gilchrist H, Jackson S, Morse L, Nicotri T, Nesbitt LT. Galli-Galli disease: A case report with review of the literature. J Am Acad Dermatol 2008;58:299-302.
[Google Scholar]
|
| 4. |
Bardach H, Gebhart W, Luger T. Genodermatosis in a pair of brothers: Dowling-Degos, Grover, Darier, Hailey-Hailey or Galli-Galli disease? Hautarzt 1982;33:378-83.
[Google Scholar]
|
| 5. |
Wu YH, Lin YC. Generalized Dowling-Degos disease. J Am Acad Dermatol 2007;57:327-34.
[Google Scholar]
|
| 6. |
Liao H, Zhao Y, Baty DU, McGrath JA, Mellerio JE, McLean WH. A heterozygous frameshift mutation in the V1 domain of keratin 5 in a family with Dowling-Degos disease. J Invest Dermatol 2007;127:298-300.
[Google Scholar]
|
| 7. |
El Shabrawi-Caelen L, Rütten A, Kerl H. The expanding spectrum of Galli-Galli disease. J Am Acad Dermatol 2007;56 5 Suppl: S86-91.
[Google Scholar]
|
| 8. |
Ostlere L, Holden CA. Dowling-Degos disease associated with Kitamura's reticulate acropigmentation. Clin Exp Dermatol 1994;19:492-5.
[Google Scholar]
|
| 9. |
Müller CS, Pföhler C, Tilgen W. Changing a concept – Controversy on the confusing spectrum of the reticulate pigmented disorders of the skin. J Cutan Pathol 2009;36:44-8.
[Google Scholar]
|
| 10. |
Verma S, Pasternack SM, Rütten A, Ruzicka T, Betz RC, Hanneken S. The first report of KRT5 mutation underlying acantholytic Dowling-Degos disease with mottled hypopigmentation in an Indian family. Indian J Dermatol 2014;59:476-80.
[Google Scholar]
|
Fulltext Views
4,913
PDF downloads
1,328





![[Figure - 1]](#fig_ijdvl_2016_82_6_720_190842_f1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2016_82_6_720_190842_f2.jpg){kind=link}
![[Figure - 3]](#fig_ijdvl_2016_82_6_720_190842_f3.jpg){kind=link}
![[Figure - 4]](#fig_ijdvl_2016_82_6_720_190842_f4.jpg){kind=link}
![[Figure - 5]](#fig_ijdvl_2016_82_6_720_190842_f5.jpg){kind=link}
![[Figure - 6]](#fig_ijdvl_2016_82_6_720_190842_f6.jpg){kind=link}
![[Figure - 7]](#fig_ijdvl_2016_82_6_720_190842_f7.jpg){kind=link}
![[Figure - 8]](#fig_ijdvl_2016_82_6_720_190842_f8.jpg){kind=link}