Translate this page into:
Benign cephalic histiocytosis
2 Department of Pathology, Necmettin Erbakan University Meram, Faculty of Medical, Konya, Turkey
Correspondence Address:
Munise Daye
Department of Dermatology, University Meram, Faculty of Medical, Konya
Turkey
| How to cite this article: Daye M, Dogan S, Mevlitoglu I, Toy H. Benign cephalic histiocytosis. Indian J Dermatol Venereol Leprol 2013;79:713-714 |
Sir,
Histiocytic skin diseases are uncommon in children. They are usually classified either as Langerhans′ cell histiocytosis (LCH) or non-LCH, based on the pathology of the proliferating histiocytes. Among the main types of non-LCH are benign cephalic histiocytosis (BCH), early juvenile xanthogranuloma (JXG), xanthoma disseminatum and generalized eruptive histiocytosis (GEH). [1],[2] Here, we describe a 14-month-old boy that had clinicopathologic features consistent with the diagnosis of BCH. A 14-month-old boy was referred to our out-patient clinic with complaint of small discrete, asymptomatic, yellow-red papules since the age of 9 months. The number of papules gradually increased over the weeks and spread to his upper trunk, back, shoulders, and upper arms. Dermatologic examination at the time of presentation demonstrated asymptomatic, multiple, scattered yellow-red colored, monomorphic papules and macules that were 2-5 mm over his cheeks, chest, upper back and arms [Figure - 1]. Hematologic and biochemical examinations, skeleton surveys, diabetes insipidus (DI), relevant antidiuretic hormone (ADH) levels, concurrent urinary osmolarity and blood biochemistry, insulin levels were normal. Histologic examination of the biopsy specimen taken from a lesion on the left shoulder showed a normal epidermis and histiocytic cells within papillary dermis without epidermotropism. In the immunohistochemistry analysis, the cells were stained positive for cluster of differentiation 68 (CD68), but negative for the S100 protein and CD1a [Figure - 2]a and b. Staining for mast cells were negative. The case has been diagnosed BCH with these clinicopathological features. BCH is a rare non-Langerhans histiocytic disorder of childhood, which presents as a self-healing eruption of head and neck. [3] Since its description by Gianotti et al., there have been 42 published cases in the English- language literature. These previous cases were thoroughly reviewed by Jih et al.; among the 40 patients they reviewed, the age at onset ranged from 2 to 66 months. [1],[3] In approximately 50% of cases, the patients are younger than 6 months. Males and females are affected equally. [1] Our case was a 14-month-old boy and his eruptions appeared firstly at the age of 9-month. Etiology of disease is still unknown. Some investigators believe that it is a variant of JXG or GEH or perhaps they all belong within the spectrum of one single disease. The clinical features are small, papules and macules that are 1-8 mm in diameter with colours ranging from yellow to red-brown distributed mainly on the head, face, neck, and shoulders. Lesions may subsequently extend over the body. [3],[4] There is no mucous membrane, acral and visceral involvement as in our case. [3] Although, systemic disease has not been associated with BCH, DI and insulin-dependent diabetes mellitus have been reported in association with BCH. [2],[4],[5] Our patient had multiple, 2-5 mm yellow-red monomorphic papules that first appeared on the face and spread over the upper part of the body and upper extremities. Hematologic and biochemical examinations, skeleton surveys, ADH hormone levels concurrent urinary osmolarity and blood biochemistry, insulin were normal. We did not find any association with other diseases in our case. BCH must primarily be differentiated from micronodular form of JXG and GEH. Lesions of JXG are not usually limited to the head and neck, but rather are more widely distributed over the entire body. Extracutaneous involvement occurs most significantly in the eyes. Histologically, abundant foamy cells and Touton giant cells can be seen in JXG. GEH is more common in adults, with a more extensive distribution of lesions, and occasional mucosal lesions. The histologic appearance may be similar to BCH. Furthermore, LCH, plane warts, urticaria pigmentosa, lichenoid sarcoidosis and multiple Spitz nevi can be confused with BCH. [3] The histopathologic hallmark of BCH is of a well-circumscribed histiocytic infiltrate in the superficial to mid-reticular dermis. This is associated with a mixed inflammatory infiltrate of lymphocytes, rarely, eosinophils. The Langerhans cell markers CD1a and S-100 protein are negative on immunohistochemical staining, whereas the macrophage/histiocytic markers (CD68 and factor XIIIa) are positive. Histopathologic and immunhistochemical staining of the case showed dermal CD1a/S-100 negative, CD68 positive histiocytic cell infiltrate. Spontaneous regression of lesions begins on an average between 8 and 48 months and complete regression occurs on an average at 50 months. [1],[3] During the follow-up time of our case, we didn′t see new lesions and there was no regression of lesions. Since BCH′s spontaneous regression is known, nature of BCH should be explained to the patient and the family. Although, no cases of BCH in association with systemic diseases have been reported, rare cases of DI or insulin-dependent diabetes mellitus in BCH patients, as well as transitions to other non-LCH disorders, have been reported. [1],[4] The cause of DI or insulin-dependent diabetes mellitus that could be seen in BCH is not known. BCH is thought to represent a reactive histiocytic process to an infectious agent. Both diseases could result from a response to a viral infection. [5] The periodic follow-up of patients′ would be advisable. [1],[4] We differentiated BCH from other diseases by clinical and immunohistopathological features of BCH. Our case has been included in periodic polyclinic follow-up. Our case is one of the rare BCH case reported from Turkey.
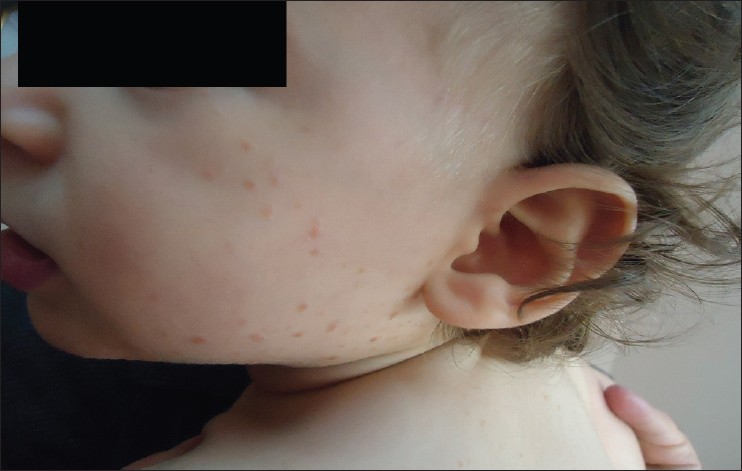 |
| Figure 1: Small, yellow-red papules and macules on face and below chin, front-upper part of trunk |
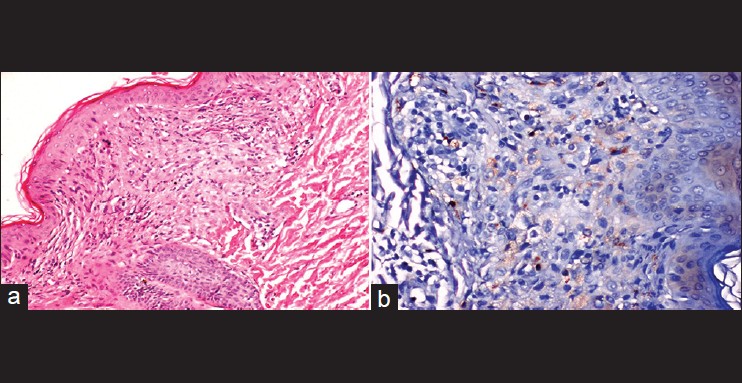 |
| Figure 2: (a) Proliferations of pleomorphic epithelioid histiocytic cells within the dermis (H and E, ×40). (b) Immunohistochemistry was positive stained for cluster of differentiation 68 (CD68) (biotin-streptavidin peroxidase system, 3,3' diaminobenzidine (DAB), ×40) |
| 1. |
Jih DM, Salcedo SL, Jaworsky C. Benign cephalic histiocytosis: A case report and review. J Am Acad Dermatol 2002;47:908-13.
[Google Scholar]
|
| 2. |
Weston WL, Travers SH, Mierau GW, Heasley D, Fitzpatrick J. Benign cephalic histiocytosis with diabetes insipidus. Pediatr Dermatol 2000;17:296-8.
[Google Scholar]
|
| 3. |
Koca R, Bektaþ S, Altinyazar HC, Sezer T. Benign cephalic histiocytosis: A case report. Ann Dermatol 2011;23:508-11.
[Google Scholar]
|
| 4. |
Kim BC, Choi WJ, Seung NR, Park EJ, Cho HJ, Kim KH, et al. A case of benign cephalic histiocytosis. Ann Dermatol 2011;23:S16-19.
[Google Scholar]
|
| 5. |
Saez-De-Ocariz M, Lopez-Corella E, Duran-McKinster C, Orozco-Covarrubias L, Ruiz-Maldonado R. Benign cephalic histiocytosis preceding the development of insulin-dependent diabetes mellitus. Pediatr Dermatol 2006;23:101-2.
[Google Scholar]
|
Fulltext Views
4,246
PDF downloads
1,955





![[Figure - 1]](#fig_ijdvl_2013_79_5_713_116750_f1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2013_79_5_713_116750_f2.jpg){kind=link}