Translate this page into:
Cutaneous plasmacytosis with mast cell infiltration
2 Skin and VD Consultant, Sankalp Hospital, Mapusa, Goa, India
Correspondence Address:
Sarina Jain
Department of Dermatology, Main Hospital Building, Ward 17-18, Ground Floor, KEM Hospital, Parel, Mumbai - 400 012, Maharashtra
India
| How to cite this article: Jain S, Hede RV, Khopkar US. Cutaneous plasmacytosis with mast cell infiltration. Indian J Dermatol Venereol Leprol 2020;86:91-95 |
Abstract
Cutaneous plasmacytosis is a rare disorder of uncertain etiology, described mainly in patients of Japanese descent. Clinically, it is characterized by multiple pigmented papules and plaques distributed primarily on the trunk. Histopathologically, it is marked by a dense dermal plasma cell infiltrate. Here, we describe a case of cutaneous plasmacytosis in a 55-year-old Indian male who presented with hyperpigmented plaques on the body. Histopathological examination revealed dense superficial and deep perivascular and periappendageal infiltrate composed mainly of plasma cells, lymphoid follicles with reactive germinal centres, perineural distribution of plasma cells, mast cell infiltration and increased dermal small blood vessels. Immunohistochemical analysis confirmed the polyclonal nature of the plasma cells. Laboratory investigations were within normal limits, except for the presence of polyclonal hypergammaglobulinemia without any M band. There was no evidence of autoimmune disease or any infection. There was no systemic involvement in this patient. The patient was diagnosed as cutaneous plasmacytosis and advised long-term follow-up. Peculiar histopathological finding in this case of cutaneous plasmacytosis was the presence of abundant mast cells in the dermis.
Introduction
Cutaneous plasmacytosis is a rare and less recognized entity. It is diagnosed on the basis of the presence of characteristic clinical and histopathological findings after excluding the secondary causes of plasma cell infiltration in the skin.[1] Here, we report a case of cutaneous plasmacytosis with mast cell infiltration in the dermis, a histopathological finding less commonly described in cutaneous plasmacytosis.
Case Report
A 55-year-old Indian male presented with asymptomatic, widespread, progressive, hyperpigmented lesions over the body for the last 6 years. He gave a history of multiple treatments taken in the form of oral prednisolone (dose 20–40 mg/day), oral antibiotics (doxycycline 100 mg/day), topical clobetasol propionate 0.05% cream, etc., with minimal relief. Cutaneous examination revealed multiple, scattered, dark brown-colored, nontender, infiltrated, nonscaly plaques and papules of varying sizes (ranging from 0.5 cm to 3 cm) and shapes (oval, elongated and irregular) distributed symmetrically onthe neck, trunk, axilla, upper arms and thighs in a Christmas tree-like pattern [Figure - 1]a and [Figure - 1]b. Face, palms and soles were sparedstroking the lesions did not induce wheal and flare response (negative Darier's sign). On general physical examination, there was no lymphadenopathy or hepatosplenomegaly. The patient was otherwise healthy with no other relevant past medical history. Our differential diagnoses were cutaneous mastocytosis, lymphoma, leukemia and secondary syphilis.
 |
| Figure 1: |
On blood investigation, his hemoglobin was 12.7 gm/dl and total leukocyte count was 6700/mm with normal differential leukocyte count. Liver and renal function tests were within normal limits. Erythrocyte sedimentation rate was normal (17 mm/1st h). Serum protein electrophoresis revealed total protein as 8.70 g/dl (normal range: 6.40–8.30 g/dl), albumin 3.97 g/dl (normal range: 3.50–5.78 g/dl), gammaglobulin 2.58 g/dl (normal range: 0.62–1.57 g/dl), A/G ratio of 0.84 (normal range: 0.90–2.00), IgG 1890 mg/dl (normal range: 700–1600 mg/dl), IgA 772 mg/dl (normal range: 70–400 mg/dl) and IgM 292 mg/dl (40–230 mg/dl), with no M spike. Hence, he had slight hyperproteinemia with polyclonal hypergammaglobulinemia. Urine analysis was within normal limits with no proteinuria. There was no measurable monoclonal Bence Jones protein in the urine. Bone marrow biopsy revealed no abnormal findings. Tests for antinuclear antibodies, rapid plasma reagin and anti-human immunodeficiency virus antibodies were negative. X-ray of the chest (posteroanterior and lateral views) and ultrasound of the abdomen and pelvis did not reveal any significant abnormal findings.
Skin biopsy from an abdominal plaque revealed epidermal acanthosis along with increased melanin pigment in the basal layer. The dermis showed superficial and deep nodular perivascular and periappendageal lymphoplasmacytic infiltrate with predominance of plasma cells [Figure - 2]a. Some of the nodules had lymphoid follicles, with reactive germinal centres, surrounded by plasma cells [Figure - 2]b. Plasma cells had small and uniform-sized nuclei [Figure - 2]c. Perineural distribution of plasma cells was also present at places [Figure - 2]d. Mast cells were found to be increased in number in the dermal interstitium, as evidenced by Giemsa stain [Figure - 2]e. A mild interstitial infiltrate consisting of lymphocytes and histiocytes was also observed. Dermal small blood vessels were also increased in number.
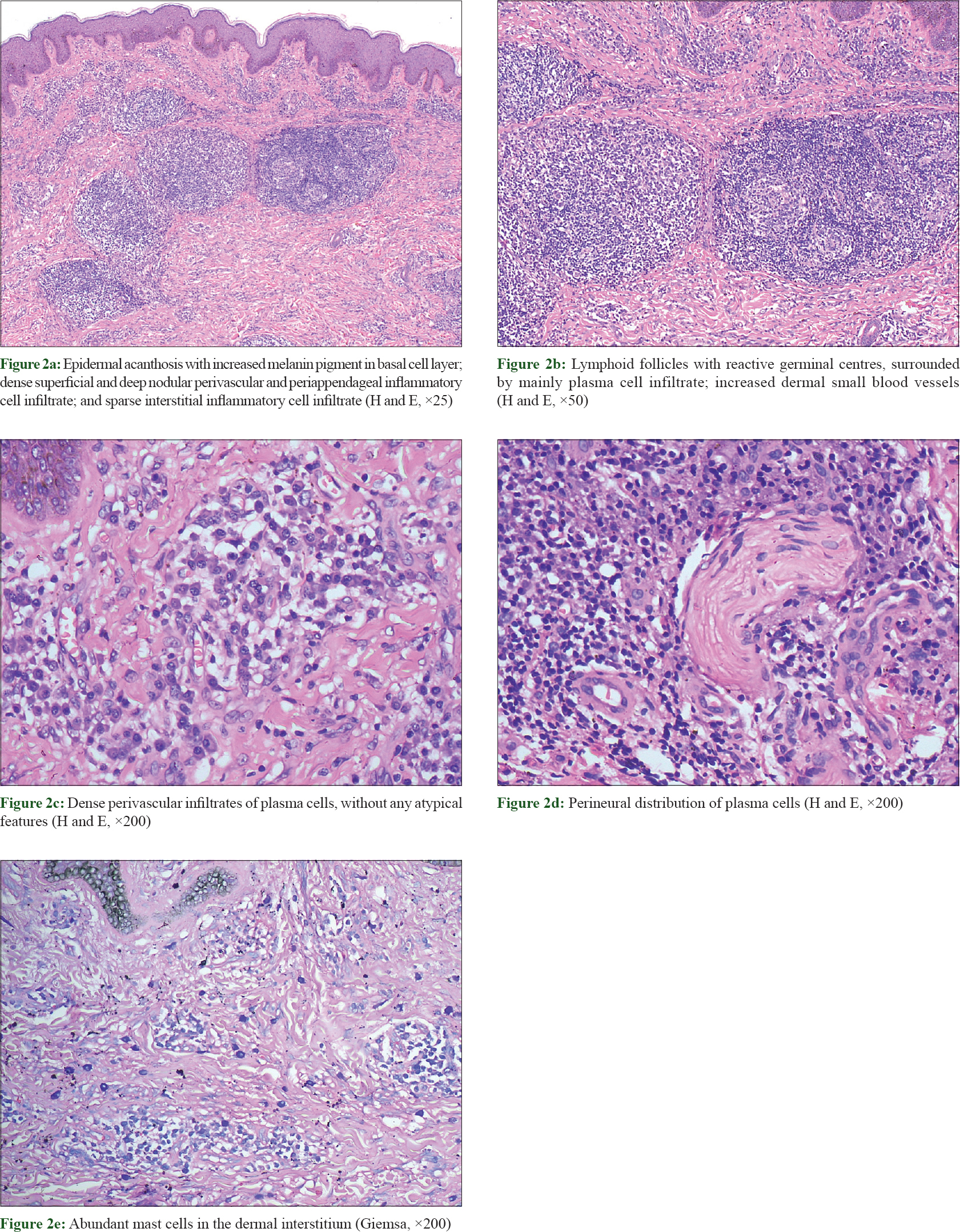 |
| Figure 2: |
Immunohistochemical examination showed plasma cells positive for CD138 [Figure - 3]a and a polyclonal staining pattern with kappa and lambda light chains in a ratio of 1:1 [Figure - 3]b and [Figure - 3]c.
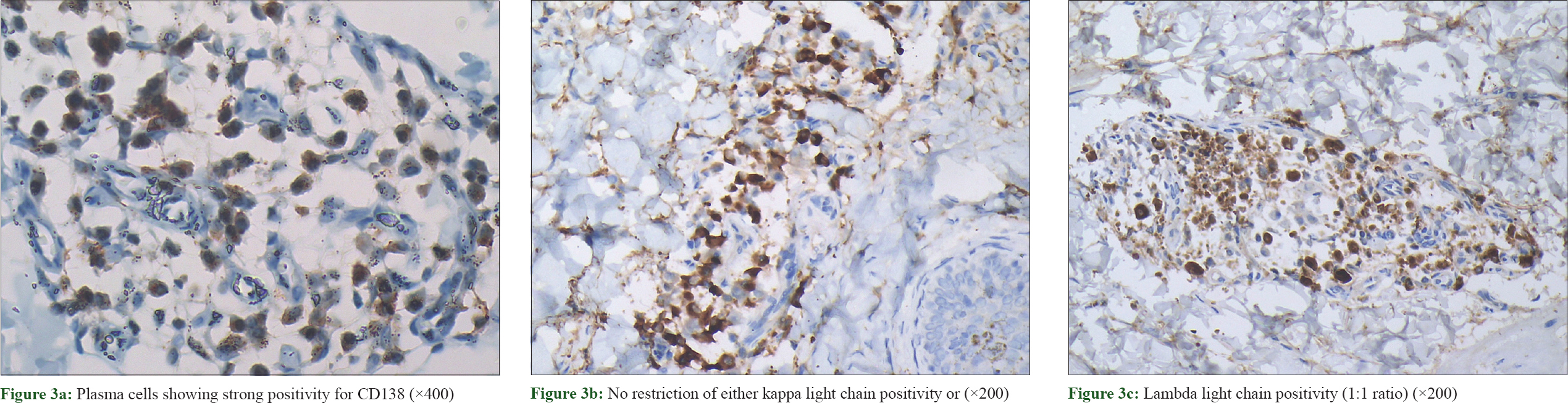 |
| Figure 3: |
Based on the above findings, cutaneous plasmacytosis was diagnosed. The patient declined any further treatment at this time. The patient was advised long-term follow-up to monitor possible development of any systemic involvement.
Discussion
Cutaneous plasmacytosis was first recognized as a distinctive dermatosis by Yashiro et al. in 1976.[1] Kimura et al. coined the term “cutaneous plasmacytosis” to describe a condition marked by the proliferation of mature plasma cells in the skin, which manifests as multiple cutaneous plaques and polyclonal hypergammaglobulinemia.[2] Recently, a more comprehensive term “cutaneous and systemic plasmacytosis” was suggested as some cases of cutaneous plasmacytosis may have occult involvement of extracutaneous areas such as lymph nodes and bone marrow.[3] The term cutaneous plasmacytosis is used when there is skin involvement only, without involvement of any extracutaneous sites.[4]
Cutaneous plasmacytosis has a characteristic clinical presentation in the form of multiple, asymptomatic, infiltrated, dark-brown, irregularly-shaped macules, plaques and flat tumors located primarily on the trunk with particular involvement of axilla. The lesions are commonly distributed in a Christmas tree-like pattern.[1] Cutaneous lesions are commonly associated with polyclonal hypergammaglobulinemia.
The most common manifestation of extracutaneous involvement is superficial lymphadenopathy. Other less frequently involved sites include bone marrow, lung, liver, spleen and kidney.[5]
Histopathologically, cutaneous plasmacytosis is marked by dense superficial and deep perivascular and periappendageal infiltrate composed mainly of plasma cells along with few lymphocytes and histiocytes. Plasma cells are mature without any evidence of atypia. Other less commonly described features include perineural distribution of plasma cells and lymphoid follicles with reactive germinal centres.[6]
Increased basal layer melanin pigmentation is a commonly described feature of cutaneous plasmacytosis, the reason for which remains unclear.
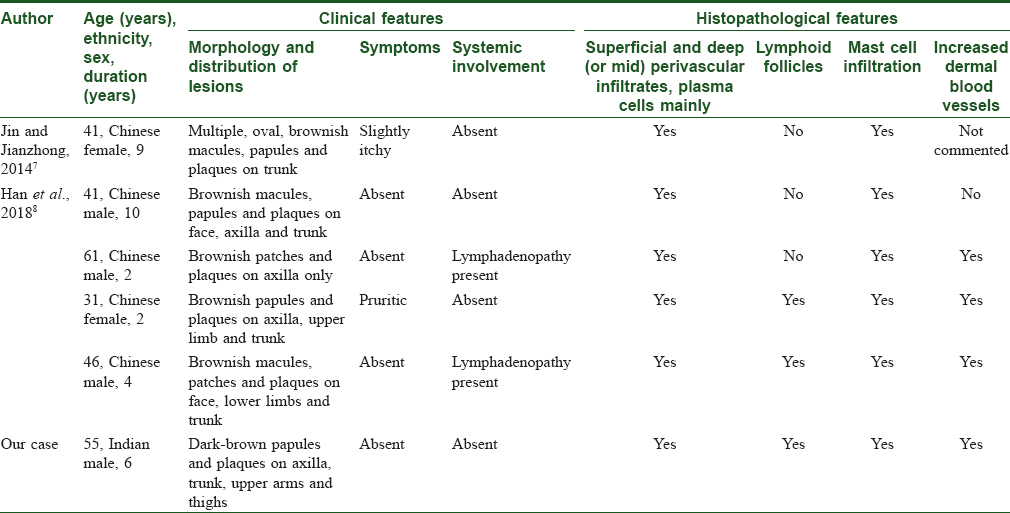
Mast cell infiltration in our case was an interesting finding. To date, only five cases of cutaneous plasmacytosis with mast cell infiltration have been documented [Table - 1].[7],[8] The significance of mast cell infiltration in cutaneous plasmacytosis is not known. Increased bone marrow mast cells are commonly found in acute leukemia and plasma cell disorders such as Waldenstrom's macroglobulinemia.[9] In 1993, Raj et al. reported two cases of acute lymphoblastic leukemia with urticaria pigmentosa-like lesions, where they found mast cell infiltration in skin lesions and bone marrow in addition to lymphoblasts.[10] Similarly, increased mast cells have also been found in skin biopsies from patients with cutaneous B- and T-cell lymphomas.[11]
Two mechanisms have been described for the presence of mast cells in lymphoproliferative and plasma cell disorders. First, the mast cell derived interleukin-6 has been shown to maintain the proliferative spur in B- and plasma cell neoplasms.[12] Second, the increased microvessel density has been correlated with increased mast cell number.[11]
Raised interleukin-6 levels are reported in some patients of cutaneous and systemic plasmacytosis.[6] We could not assess our patient's serum interleukin-6 level due to the unavailability of this test in our institution. Increased dermal small vessels were found in our case, and similar finding has been previously described in three patients by Han et al. [Table - 1]. These findings suggest the possible role of mast cells in cutaneous plasmacytosis. The presence of mast cells in cutaneous plasmacytosis may have treatment implications. The agents that interfere with interleukin-6 activity can be potential therapeutic agents. Thalidomide has been used and found to be effective in a case of cutaneous and systemic plasmacytosis based on the same mechanism.[13] Multicentric plasma cell variant of Castleman's disease is a rare type of reactive lymphadenopathy characterized by systemic involvement in the form of fever, cytopenias, thrombocytosis, hypergammaglobulinemia, splenomegaly, hepatomegaly and pulmonary involvement. Rarely, cutaneous involvement occurs in multicentric plasma cell variant of Castleman's disease with histopathological features of dermal and subcutaneous nodular plasma cell infiltrates, vascular proliferation and abortive germinal centres. Increased serum interleukin-6 levels have also been frequently seen in multicentric plasma cell variant of Castleman's disease.[14] Based on these findings, some authors have suggested “cutaneous and systemic plasmacytosis” to be a variant of multicentric plasma cell variant of Castleman's disease.
Since our case did not have any lymphadenopathy or any other systemic involvement, it fits better with cutaneous plasmacytosis.
Cutaneous plasmacytosis has a chronic benign course with an overall favorable prognosis. There is no standard effective treatment for this condition, and different treatment options have been tried with limited success.[5]
Here, we documented a case of cutaneous plasmacytosis in a 55-year-old Indian male. Our case represents one of the less frequently described histopathological manifestations of cutaneous plasmacytosis in the form of presence of numerous mast cells, increased dermal small blood vessels, lymphoid follicles with reactive germinal centres and perineural distribution of plasma cells along with classical dense dermal plasma cell infiltrates. This case expands the histopathological spectrum of this recently recognized enigmatic entity.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form, the patient has given his consent for his images and other clinical information to be reported in the journal. The patient understands that name and initials will not be published and due efforts will be made to conceal identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
| 1. |
Lu PH, Shih LY, Yang CH, Kuo TT. Cutaneous plasmacytosis: A clinicopathologic study of 12 cases in Taiwan revealing heterogeneous underlying causes. Int J Dermatol 2015;54:1132-7.
[Google Scholar]
|
| 2. |
Kimura K, Fujita H, Ishihama S. Cutaneous plasmacytosis. Rinsho Derma 1983;25:1045-50.
[Google Scholar]
|
| 3. |
Leonard AL, Meehan SA, Ramsey D, Brown L, Sen F. Cutaneous and systemic plasmacytosis. J Am Acad Dermatol 2007;56:S38-40.
[Google Scholar]
|
| 4. |
Lee SH, Yoo CY, Jung JH, Yoo JY, Kang SJ, Kang CS. Systemic plasmacytosis – A case report with a review of the literature. Korean J Pathol 2011;45:632-8.
[Google Scholar]
|
| 5. |
Dhar S, Liani L, Patole K, Dhar S. Cutaneous plasmacytosis: A rare entity with unique presentation. Indian J Dermatol Venereol Leprol 2017;83:673-6.
[Google Scholar]
|
| 6. |
Honda R, Cerroni L, Tanikawa A, Ebihara T, Amagai M, Ishiko A, et al. Cutaneous plasmacytosis: Report of 6 cases with or without systemic involvement. J Am Acad Dermatol 2013;68:978-85.
[Google Scholar]
|
| 7. |
Jin W, Jianzhong Z. Mast cell infiltration in a patient with cutaneous plasmacytosis. Austin J Dermatol 2014;1:1012.
[Google Scholar]
|
| 8. |
Han XD, Lee SS, Tan SH, Chong WS, Ng SK, Ooi MG, et al. Cutaneous plasmacytosis: A clinicopathologic study of a series of cases and their treatment outcomes. Am J Dermatopathol 2018;40:36-42.
[Google Scholar]
|
| 9. |
Prokocimer M, Polliack A. Increased bone marrow mast cells in preleukemic syndromes, acute leukemia, and lymphoproliferative disorders. Am J Clin Pathol 1981;75:34-8.
[Google Scholar]
|
| 10. |
Raj S, Khopkar U, Wadhwa SL, Kapasi A. Urticaria-pigmentosa-like lesions in acute lymphoblastic leukaemia (2 cases). Dermatology 1993;186:226-8.
[Google Scholar]
|
| 11. |
Rabenhorst A, Schlaak M, Heukamp LC, Förster A, Theurich S, von Bergwelt-Baildon M, et al. Mast cells play a protumorigenic role in primary cutaneous lymphoma. Blood 2012;120:2042-54.
[Google Scholar]
|
| 12. |
Merluzzi S, Frossi B, Gri G, Parusso S, Tripodo C, Pucillo C, et al. Mast cells enhance proliferation of B lymphocytes and drive their differentiation toward IgA-secreting plasma cells. Blood 2010;115:2810-7.
[Google Scholar]
|
| 13. |
Fang S, Shan K, Chen AJ. Cutaneous and systemic plasmacytosis on the face: Effective treatment of a case using thalidomide. Oncol Lett 2016;11:1923-5.
[Google Scholar]
|
| 14. |
Klein WM, Rencic A, Munshi NC, Nousari CH. Multicentric plasma cell variant of Castleman's disease with cutaneous involvement. J Cutan Pathol 2004;31:448-52.
[Google Scholar]
|
Fulltext Views
3,174
PDF downloads
1,524





![[Figure - 1]](#fig_ijdvl_2020_86_1_91_238510_f1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2020_86_1_91_238510_f2.jpg){kind=link}
![[Figure - 3]](#fig_ijdvl_2020_86_1_91_238510_f3.jpg){kind=link}
![[Table - 1]](#tbl_ijdvl_2020_86_1_91_238510_t4.jpg){kind=link}