Translate this page into:
Epidermolysis bullosa: Where do we stand?
Correspondence Address:
Rashmi Sarkar
Department of Dermatology and Venereology, Maulana Azad Medical College and Lok Nayak Hospital, New Delhi 110 092
India
| How to cite this article: Sarkar R, Bansal S, Garg VK. Epidermolysis bullosa: Where do we stand?. Indian J Dermatol Venereol Leprol 2011;77:431-438 |
Abstract
Epidermolysis bullosa, a genetically determined skin fragility disorder can severely incapacitate the life of the afflicted patient. Although the clinical features are multiple and varied, treatment still remains a major challenge. There have been major changes in the classification of the disease recently. Although there is still a long way to go, good nursing care, and gene therapy could possibly significantly alleviate the suffering of the patients in the future.Introduction
Epidermolysis bullosa (EB) comprises a group of genetically determined skin fragility disorders, which are characterized by blistering of the skin and mucosa, in response to little or no apparent trauma. These disorders represent heterogeneous phenotypes and are associated with a variable range of complications, from localized skin fragility to neonatal death. [1] However, the term "epidermolysis" is not correct as epidermal disruption is not the primary change in two of the main categories of EB. [2],[3] This complex and heterogeneous group is classified on the basis of the mode of inheritance, clinical, laboratory and epidemiological studies into three major forms: EB simplex (EBS), junctional EB (JEB), and dystrophic EB (DEB).
Epidemiology
The most accurate epidemiological data are derived from the National EB registry project from USA and also from Scotland. [1],[4] According to the National EB registry project from USA, the incidence and prevalence of EB are estimated to be 19.60 per million live births and 8.22 per million population, respectively. The incidence and prevalence rates of EB simplex are 10.75 and 4.65, of junctional EB are 2.04 and 0.44, and dystrophic EB dominant type 2.86 and 0.99 and recessive dystrophic EB 2.04 and 0.92, respectively. Whereas, according to the epidemiological data from Scotland, the incidence of EBS in 2001 was 33.2 per million and the incidence between 1960 and 1999 was 34.4 per million live births. [4] For junctional EB, the Scottish prevalence was 0.3 cases per million and an incidence of 3.2 new cases per million live births and for dystrophic EB, the prevalence is 24.6 cases per million and the incidence 26.4 new cases per million live births. But, the overall worldwide data suggests that there is no gender, racial, or geographical predilection of EB, although there is a possibility of underestimating the clinically milder forms of EB.
Classification
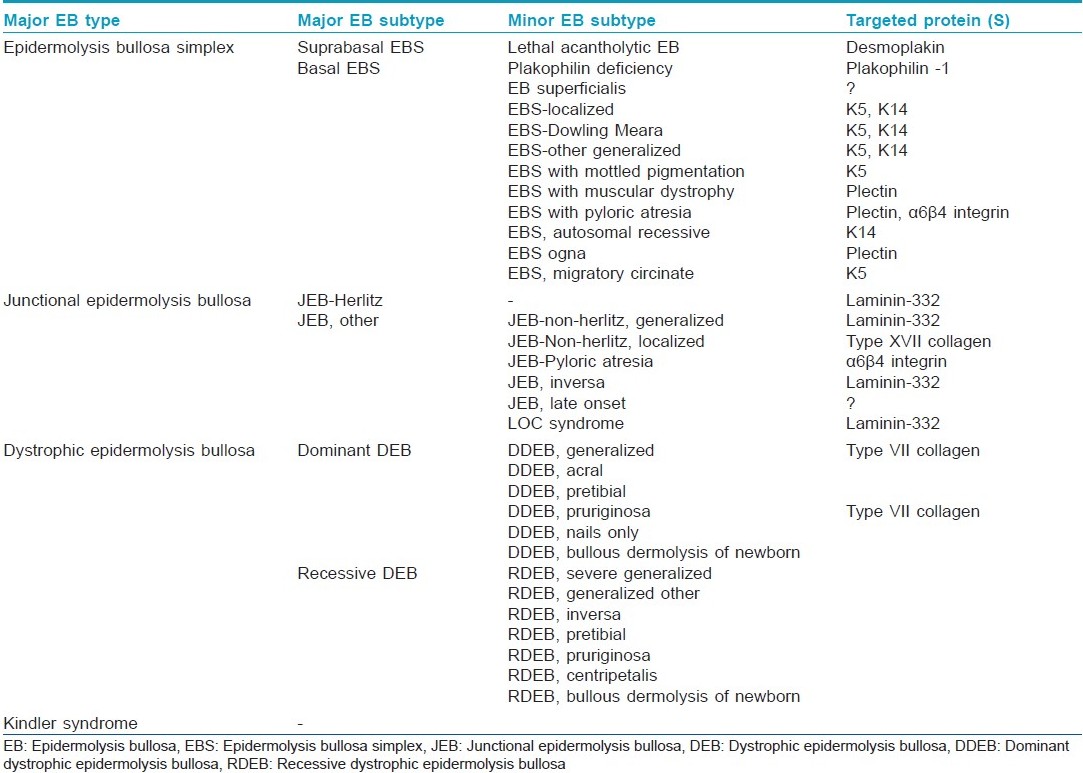
The most recent classification of EB has been proposed by the third international consensus meeting held in Vienna in 2007. [5] In this classifications, attempts were made to eliminate eponyms, streamline it, and add a fourth group to the major EB types, the mixed or Kindler Syndrome. The patients are then separated by major and minor EB subtypes [Table - 1].
Pathogenesis
With the help of immnunohistochemistry, the major epidermolysis bullosa genes identified are those that encode keratins 5 and 14 in EBS. [6] In JEB the underlying defect lies in the hemidesmosomes which tend to be sparse and very small, especially in the more severe forms of the disease and the majority of mutations have been found in the LAMB3 gene. [7],[8] Immunofluorescence studies have also shown reduced staining of another hemidesmosome-anchoring filament component, BPAG2, or collagen 17 in the skin of patients with generalized atrophic benign EB (GABEB), suggested that mutations in the BPAG2 gene might underlie this condition. [9] DEB is caused by mutations in a single gene, COL7A1, encoding the anchoring fibril protein, the type 7 collagen. Quantitative electron microscopy and immune-electron microscopy have shown complete absence of anchoring fibrils in severe forms of dystrophic EB. [10] In the milder or more localized form of recessive dystrophic EB, immunoreactivity is present but often attenuated. Currently, more than 1000 mutations, encompassing more than 10 distinct structural genes expressed within the cutaneous BMZ have now been documented for epidermolysis bullosa. [6] The compartmentalized expression of these genes within the cutaneous BMZ, the types and combinations of the mutations and their consequences at the mRNA and protein levels, when combined with the patient′s genetic background and environmental factors, can account for the severity in EB. [11] In fact, Kindler syndrome has now been included as the fourth major EB type, according to the leading authorities. [7]
Clinical Features
The major clinical types of epidermolysis bullosa and their differential diagnosis are shown in [Table - 2].

Epidermolysis Bullosa Simplex
It is the most frequent form of EB.
Clinical subtypes of epidermolysis bullosa simplex
Epidermolysis bullosa simplex, Weber--Cockayne type
Blisters are rarely present at birth and usually begin with crawling. Blisters are usually confined to the hands and feet, but can occasionally occur anywhere if trauma is significant [Figure - 1]. Symptoms are worse in warm weather and worsen with sweating. A study has demonstrated reduced thermal stability of the keratin filaments in the cell lines of patients derived from both Weber-Cockayne and Dowling-Meara forms of EBS. [12] Healing occurs without scarring or milia formation. Oral mucosa, teeth, and hair are normal.
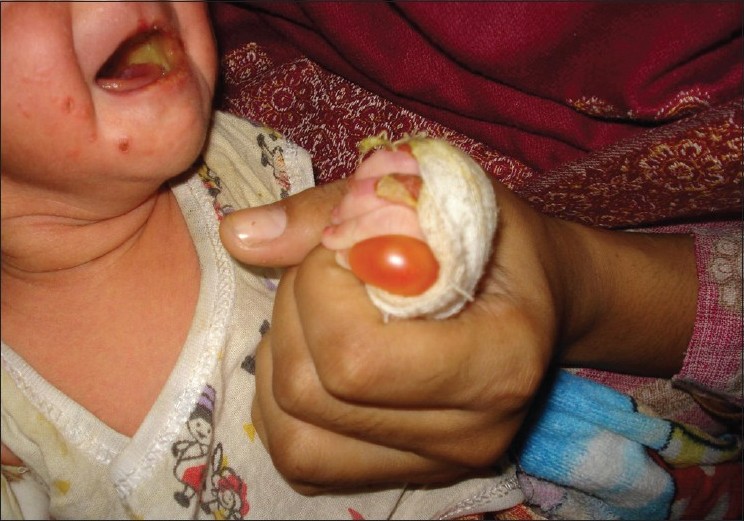 |
| Figure 1: Blisters on parts exposed to trauma of a neonate of epidermolysis bullosa simplex |
Epidermolysis bullosa simplex, Dowling--Meara type
Onset is usually at birth with widespread and severe blistering. Sometimes, multiple grouped clumps of small blisters occur which are typical of the disease hence the name EB herpetiformis. Mucosal involvement may interfere with feeding. Healing may lead to localized atrophic scarring and/or milia formation. Progressive hyperkeratosis of the palms and soles begins in childhood and may be the major complaint of affected individuals in adult life. [13] Nail dystrophy and shedding of nails has been reported. In general, improvement occurs with age.
Epidermolysis bullosa simplex, Koebner type
It is distinguished from EBS-Weber-Cockayne by its more widespread involvement, but misdiagnosis is common. In infancy, blistering commonly occurs on the occiput, back, and legs, while in childhood, hands and feet are mainly affected.
Less common forms of epidermolysis bullosa simplex
EBS with mottled pigmentation was first reported in 1979. [14] Hallmark of this subtype of EB is the presence of discrete pigmented macules 2--5 mm in diameter, which usually appear in infancy and persist throughout life. [15] The pigmentation has a predominance for the trunk and large skin folds. Histology shows increased melanin content in dermal melanophages. A recent study has suggested a possible molecular mechanism by which the P25L substitution in the nonhelical V1 domain of K5 usually found in these patients causes this unusual pigmentation. [16]
EBS with neuromuscular disease is transmitted as an autosomal recessive trait, unlike other forms of EBS. The ultrastructural level of cleavage begins at a very low level in the basal cells, just above the hemidesmosomes. Immunofluorescence microscopy of skin from affected individuals has shown diminished or absent staining for plectin. Homozygous mutations in the PLEC1 gene have been reported. The disease has been reported in association with neuromuscular diseases such as muscular dystrophy, myasthenia gravis, and spinal muscular atrophy. The widespread blistering is associated with atrophic scarring, milia, and nail dystrophy.
EB-Ogna another rare form of EBS was observed in one Norwegian and one German family, and is caused by a site-specific missense mutation within the rod domain of PLEC1. [17] It is distinguished by a generalized bruising tendency, hemorrhagic bulla, and onychogryphotic great toe nails.
EBS-superficialis is a very rare subset of EBS, described in two families by Fine et al. in 1989. Superficial erosions rather than intact blisters are the hallmark. A recent study has demonstrated a mutation in the COL7A1 gene in one kindred with EB Superficialis. [18]
Junctional Epidermolysis Bullosa
Junctional Epidermolysis Bullosa (JEB) encompasses a group of autosomal recessive disorders characterized by blister formation at the level of lamina lucida.
Clinical subtypes of junctional epidermolysis bullosa
Herlitz junctional epidermolysis bullosa
Blisters are present at or soon after birth. Nonhealing crusted erosions with significant granulation tissue are the hallmark which appears characteristically around the nose, mouth, ears, and tips of the fingers and toes. [19] There is extensive loss of blood, fluid, and protein. Many infants die early in infancy as a result of electrolyte imbalance and overwhelming infection. In those who survive, healing generally results in atrophic scarring. Mucosal involvement of the mouth, upper respiratory tract, esophagus, bladder, urethra, and corneas has also been reported leading to hoarseness, stridor, urinary retention, urinary tract infections, and eventual renal compromise. Teeth show abnormal enamel formation with normal dentine, leading to malformation, pitting, and premature loss. [20]
Non-Herlitz junctional epidermolysis bullosa or generalized atrophic benign epidermolysis bullosa (GABEB or Hinter Wolf type)
The condition was first described by Hashimoto et al. in 1976. This is less severe than the classic form of JEB, although esophageal stricture, laryngeal involvement, corneal ulcers and urethral stricture all have been reported. The lesions usually heal with atrophic scarring which can easily be mistaken for scarring seen in dystrophic EB. Varying degrees of alopecia, onychodystrophy, tooth pitting as well as increased chances of squamous cell carcinoma remain hallmarks of this type of JEB. [21]
Less common forms of junctional epidermolysis bullosa
JEB with pyloric atresia is a rare disorder in which the true level of blistering has been found in the cytoplasm of basal keratinocytes rather than in the lamina lucida. Blistering is usually severe and present at birth following a pregnancy complicated by polyhydroamnios. Atrophic scarring, hypoplastic teeth and dystrophic nails all have been reported. [22]
EB progressiva is another rare form of JEB characterized by progressive atrophic changes leading to early loss of finger print patterns and mild finger contractures. Electron microscopy studies have shown a normal dermo--epidermal junction zone, including normal hemidesmosomes. The molecular basis is unknown.
Cicatricial JEB: Three cases of this new type of JEB were described by Haber et al. in 1985 characterized clinically by blisters that healed with scarring, syndactyly, contractures and stenosis of the anterior nares. The cases were believed to represent a dystrophic type of epidermolysis bullosa (EB) clinically and emphasized the necessity for electron microscopy of the skin in all EB patients with dystrophic features in order to make a definite diagnosis. However, based on recent studies, this phenotype has been clubbed under the heading of Non-Herlitz JEB.
Inverse JEB is another rare form characterized by lesions chiefly affecting the groin, perineum and axillae. Healing results in small atrophic white streaks.
Dystrophic Epidermolysis Bullosa
Clinical subtypes of dystrophic epidermolysis bullosa
Recessive dystrophic epidermolysis bullosa
Hallopeau-Siemens type recessive dystrophic epidermolysis bullosa
Blisters appear at birth or in the neonatal period. Blisters affect the whole body and may be hemorrhagic. Scarring and milia are a constant feature leading to disfigurement. Scarring pseudosyndactyly of the hands and feet fuses the digits into "mitten" hands and feet with severe loss of function. Mucosal involvement is usually severe and appear early in life. Oral involvement may lead to ankyloglossia and microstomia, which along with painful esophageal erosions impair food intake. [23],[24] Anal erosions can cause severe constipation. Corneal erosions can lead to scarring and loss of vision. General physical development is retarded and sexual maturation is generally delayed. Studies have shown reduced levels of vitamins and trace metals in the blood and impairment of immune function. Development of squamous cell carcinomas has been reported. [25] Other complications include osteoporosis, renal amyloidosis, pulmonary amyloidosis, and dilated cardiomyopathy.
Non-Hallopeau-Siemens type recessive dystrophic epidermolysis bullosa
It shares all the clinical features with the more severe type of recessive dystrophic epidermolysis bullosa (RDEB), but lacks the severe, mutilating scarring seen in classical RDEB.
In another variant of RDEB known as RDEB inversa, blistering and skin atrophy occurs on the neck, thighs, groins and axillae while no changes are observed on the hands, feet, elbows, or knees.
Dominant dystrophic epidermolysis bullosa
Classical dominant dystrophic epidermolysis bullosa (Cockayne--Touraine type)
It is a mild form of DEB, where blistering is often limited to the bony prominences, but nonetheless heals with scarring and milia formation. Dystrophic nails are the hallmark especially in adults, who lack the characteristic scarring. [26],[27] A variant of classical dominant dystrophic epidermolysis bullosa (DDEB) was described by Pasini in 1928, distinguished by the presence of numerous white papules called as albopapuloid lesions. The histogenesis of albopapuloid lesions remains unclear.
Less common forms of dominant dystrophic epidermolysis bullosa
Congenital localized absence of skin (Bart type) is characterized by localized absence of skin over lower legs along with cutaneous and mucosal blisters. However, absence of the skin can be seen in any of the three major types of EB, and hence should be regarded as a manifestation of EB, rather than a separate entity.
Pretibial DEB and DEB pruriginosa is a newly characterized variant of dystrophic EB. The overriding symptom is severe pruritis. The cause of pruritis is unknown, however a number of patients have raised blood levels of immunoglobulin E.
Transient bullous dermolysis of the newborn is a rare form of dystrophic epidermolysis bullosa (DEB) that presents with neonatal skin blistering but which usually improves markedly during early life or even remits completely. Skin biopsies reveal abnormal intraepidermal accumulation of type VII collagen which results in poorly constructed anchoring fibrils and a sublamina densa plane of blister formation. [28] The reason for the spontaneous clinical improvement is not known, but there is a gradual recovery in type VII collagen secretion from basal keratinocytes to the dermal-epidermal junction, with subsequent improvement or correction of anchoring fibril morphology.
Laboratory Diagnosis
Diagnostically, EB remains a challenge. The definitive diagnosis of inherited EB is made with transmission electron microscopy (TEM), immunofluorescence antigen mapping (IF), and EB related monoclonal antibody testing as well as mutational analysis. In order to make a correct diagnosis, it is most important that the skin biopsy should be performed properly, as described by Intong and Murell. [29]
The best areas to take skin biopsies in EB patients are fresh blisters (less than 1-hour old) or an unaffected area of the skin preferably adjacent to the site where the patient usually gets blisters. Before performing the biopsy, attempts should be made at inducing microscopic cleavage except in the cases of severe junctional and recessive dystrophic EB where there is such inherent mechanical fragility that it is easy to demonstrate skin cleavage planes even with routine punch biopsy. The biopsy site should be cleaned in a sterile manner and encircled. The method of rubbing consists of applying firm downward pressure with an eraser or pencil eraser and then laterally in a rotary fashion (at least 180 each way) up to even 20 times in newborns and infants. Although the area may turn red, wait for at least five minutes before taking a skin biopsy for a blister to develop microscopically. However, precaution must be taken not to tear or peel off the skin.
The selected area is then cleaned and draped and anaesthetized with 1% lignocaine or a cream of a eutectic mixture of local anesthetics under occlusion to the site for 2 h beforehand in a child patient. [30] A 3 mm punch biopsy is then performed using a twisting movement from the rubbed area and then placed in Michel′s solution or liquid nitrogen or normal saline for transport. A 2 mm punch biopsy from the same area, should be taken and placed in 2.5% glutaraldehyde solution for electron microscopy. A sample can also be taken from an unaffected area. Suturing and dressing of the biopsy area is then done.
TEM was the first tool used to achieve a correct diagnosis and classification of the disease and has remained the gold standard laboratory test for several years. [1] However, it is time consuming, expensive, and operator dependent. Immunofluorescence mapping is as diagnostically reliable as TEM and is based on the detection of structural proteins of keratinocytes or of the dermo-epidermal junction using poly or monoclonal antibodies. It is used preferentially because of rapidity of results, ease in performance, and the ability to use the same specimens for study with monoclonal antibodies. According to the proteins targeted for mutational analysis, IgG antibodies are used against cytokeratin 5, cytokeratin 14, plectin, α6, and β4 integrin, type XVII collagen, laminin 332, type VII collagen and antitype IV collagen in cases of DEB.
In patients of EBS, cytokeratin 5 and 14, plectin, α6 and β4 integrin is involved in the pathology and the split is formed within the epidermis by cytolysis of basal keratinocytes. [31] In JEB, the main target proteins are type XVII collagen and laminin 332 which are expressed normally or are reduced or absent and in a blister, they appear on the roof or floor depending on the subtype of JEB. The DEB subtypes are mostly caused by VII collagen mutations. In severe generalized recessive DEB, type IV collagen antibody is used to visualize the level of blister formation, and a reaction on the blister roof indicates dermolytic blistering confirming DEB.
Mutational analysis is the ultimate means of determining the mode of inheritance and the precise site and type of molecular mutation. Prenatal and preimplantation diagnosis can also be performed, so that genetic counseling can be done. Mutational and linkage analysis are done using DNA from the proband and his or her relatives, [32] by drawing blood samples or taking buccal swabs. Methods as restriction enzyme digestion and allele specific amplification applied to detection of recurrent mutations speed up gene screening in new patients. [33],[34]
Management
In spite of extensive research on the molecular mechanisms and clinical manifestations of various forms of EB, a definitive treatment for the disease is still far from reality. The treatment can be best divided into general management and newer molecular therapies.
General
Avoidance of provoking factors for blistering remains the mainstay of management. Heat and humidity lower the threshold for blistering in patients with EB simplex, and therefore measures to reduce both these factors are important.
A key to successful management is expert nursing care. Nursing the babies on thick foam pads protects them from undue trauma induced blistering. Special precautions need to be taken for older children in the use of adhesive tapes, sphygmomanometer cuffs, tourniquets and other instruments that cause shearing of skin or mucous membranes.
Further management needs to be undertaken in a multidisciplinary set-up keeping in mind the multisystem manifestations of the disease. The erosions should be cleaned with sterile normal saline and covered with nonadherent dressings. Topical antibiotics are generally avoided because of the risk of emergence of antibiotic resistant bacteria. Oral and dental care should commence as soon as tooth eruption begins. [35] Good oral hygiene, treatment of caries and reconstructive procedures in the form of crown placements and tooth implants should be applied to maintain function. Esophageal strictures require balloon dilatation while constipation needs to be managed with adequate fiber diet and stimulant laxatives.
Early exercises and physiotherapy may help reduce the severity of contractures, which are almost inevitable and surgical release of the contracted fingers then needs to be done involving skin grafting and postoperative splinting.
Vitamins and trace metal deficiencies are frequent even in patients receiving gastrostomy feeding. [36] Regular nutritional evaluation is therefore necessary followed by dietary advice and adequate supplementation.
Patients with EB frequently experience severe pain unresponsive to conventional treatment. Recent experience has suggested the use of topical opiates in treatment of pain reducing the need for powerful systemic analgesia. Amitriptylline is useful in pain management in both adults and children.
As far as systemic treatment is concerned, none of the agents studied so far have proved to be effective in controlling blistering in patients with EB. While systemic corticosteroids are generally avoided because of the high risk of complications, a randomized controlled trial with phenytoin no more supports its use in patients with RDEB as no significant effect was noted over placebo in this study. [37] The trials with tetracyclines have failed to provide any conclusive evidence owing to the high drop-out rates. [38] Other systemic agents tried in small case series include minocycline, vitamin E, cyclosporine and retinoids; currently, there is no reliable clinical trial evidence for interventions in EB. [39]
Molecular therapies
Gene therapy is the ultimate goal which is likely to become a reality in the near future. In principle, there are two broad approaches to gene therapy-ex vivo and in vivo. Ex vivo gene therapy primarily revolves around the use of cultured cells, such as keratinocytes, obtained from the affected individuals; these cells are propagated and transduced in culture to re-express the gene that is defective in the patients. In vivo gene therapy uses the introduction of the transgene directly into the target tissue. This can be approached by either direct injection of the genetic material into the skin or mucous membranes, utilizing biolistic particle bombardment ("gene gun"), or topical application mediated by physico-chemical means, including liposomes, in vivo electroporation, etc. The objective is to introduce one normal allele into somatic cells that would replace one of the two mutant alleles in recessive forms of EB. For autosomal dominant forms, the objective is to inactivate or nullify the action of the mutant allele rather than introduction of the normal allele.
Michele De Luca reported the first-ever successful gene therapy for EB. [40] In a phase I/II clinical trial, he and his colleagues have "cured" areas of skin on the anterior thighs of a single, adult male patient with non-Herlitz junctional EB by transplanting several genetically modified epidermal sheets grown in culture onto both legs (a total of about 500 cm 2 ). The epidermal sheets were grown from the patient′s own laminin 5-3-chain-deficient epidermal stem cells taken from palm biopsies and transfected ex vivo with a retroviral vector expressing normal laminin 5-3. Following on this "proof of principle," De Luca′s team is planning the step-by-step replacement of a large proportion of this patient′s skin surface with genetically modified skin; and also a Europe-wide clinical trial with further patients. Bone marrow stem cell transplantation has been successfully used in the treatment of recessive dystrophic epidermolysis bullosa.
Recent advances in the molecular genetics has made prenatal and preimplantation genetic diagnosis possible broadening the available options in management of patients affected by EB.
Dunnill et al.[41] has performed a successful prenatal diagnosis using intragenic and flanking COL7A1 markers in a pregnancy at risk for recessive DEB.Recently, chorionic villi immunofluorescence examination has successfully been used for prenatal diagnosis of inherited epidermolysis bullosa with pyloric atresia. [42]
| 1. |
Fine JD, Johnson LB, Suchindran C, Moshell A, Gedde-Dahl T, Jr. The epidemiology of inherited epidermolysis bulllosa: Findings in the US, Canadian and European study populations. In: Fine JD, Bauer EA, Mc Guire J, Moshell A, editors. Clinical, epidemiological and laboratory advances, and the findings of the national epidermolysis bullosa registry. Baltimore: John′s Hopkings university press; 1999. p. 101-13.
[Google Scholar]
|
| 2. |
Briggaman RA. Hereditary epidermolysis bullosa with special emphasis on newly recognized syndromes and complications. Dermatol Clin 1983;1:263-80.
[Google Scholar]
|
| 3. |
Solovan C, Ciolan M, Olariu L. The biomolecular and ultrastructural basis of epidermolysis bullosa. Acta Dermatovenerol Alp Panonica Adriat 2005;14:127-35.
[Google Scholar]
|
| 4. |
Horn HM, Priestley GC, Eady RA, Tidman MJ. The prevalence of epidermolysis bullosa in Scotland. Br J Dermatol 1997;136:560-4.
[Google Scholar]
|
| 5. |
Fine JD, Eady RA, Bauer EA, Bauer JW, Bruckner-Tuderman L, Heagerty A, et al. The classification of inherited epidermolysis bullosa (EB): Report of the Third International Consensus Meeting on Diagnosis and Classification of EB. J Am Acad Dermatol 2008;58:931-50
[Google Scholar]
|
| 6. |
Mc Grath JA, Mc Millan JR, Dunnil M, Pulkkinen L, Christiano AM, Rodeck CH, et al. Genetic basis of lethal junctional epidermolysis bullosa in an affected fetus: Implications for prenatal diagnosis in one family. Prenat Diagn 1995;15:647-54.
[Google Scholar]
|
| 7. |
Eady RAJ, McGrath JA, McMillan Jr. Ultrastructural genetic disorder of skin: The dermal-epidermal junction. J Invest Dermatol 1994;103:13S-18S.
[Google Scholar]
|
| 8. |
Christiano AM, Uitto J. Molecular complexity of the cutaneous basement membrane zone. Exp Dermatol 1996;5:1-11.
[Google Scholar]
|
| 9. |
Jokman MF, de Jong MC, Heeres K, Pas HH, Vander Meer JB, Owaribe K, et al. 180-kD bullous pemphigoid antigen (BP180) is deficient in generalized atrophic benign epidermolysis bullosa. J Clin Invest 1995;95:1345-52.
[Google Scholar]
|
| 10. |
McGrath JA, Ishida-Yamamotoa, O′Grady A, Leigh IM, Eady RA. Structural variation in anchoring fibrils in dystrophic epidermolysis bullosa: Correlation with type VII collagen expression. J Invest Dermatol 1993;100:366-72.
[Google Scholar]
|
| 11. |
Uitto J, Pulkkinen L, Ringpfeil F. Progress in molecular genetics of heritable skin diseases: The paradigms of epidermolysis bullosa and pseudoxanthoma elasticum. J Investig Dermatol Symp Proc 2002;7:6-16.
[Google Scholar]
|
| 12. |
Morley SM, Dundas SR, James JL, Gupta T, Brown RA, Sexton CJ, et al. Temperature sensitivity of the keratin cytoskeleton and delayed spreading of keratinocyte lines derived from EBS patients. J Cell Sci 1995;108:3463-71.
[Google Scholar]
|
| 13. |
Shemanko CS, Mellerio JE, Tidman MJ, Lane EB, Eady RA. Severe palmo-plantar hyperkeratosis in Dowling-Meara epidermolysis bullosa simplex caused by a mutation in the keratin 14 gene (KRT14). J Invest Dermatol 1998;111:893-5.
[Google Scholar]
|
| 14. |
Fischer T, Gedde-Dahl T Jr. Epidermolysis bullosa simplex and mottled pigmentation: A new dominant syndrome. I. Clinical and histological features. Clin Genet 1979;15:228-38.
[Google Scholar]
|
| 15. |
Irvine AD, Rugg EL, Lane EB, Hoare S, Peret C, Hughes AE, et al. Molecular confirmation of the unique phenotype of epidermolysis bullosa simplex with mottled pigmentation. Br J Dermatol 2001;144:40-5.
[Google Scholar]
|
| 16. |
Uttam J, Hutton E, Coulombe PA, Anton-Lamprecht I, Yu QC, Gedde-Dahl T Jr, et al. The genetic basis of epidermolysis bullosa simplex with mottled pigmentation. Proc Natl Acad Sci U S A 1996;93:9079-84.
[Google Scholar]
|
| 17. |
Koss-Harnes D, Hoyheim B, Anton-Lamprecht I, Gjesti A, Jorgensen RS, Jahnsen FL, et al. A site-specific plectin mutation causes dominant epidermolysis bullosa simplex Ogna: Two identical de novo mutations. J Invest Dermatol 2002;118:87-93.
[Google Scholar]
|
| 18. |
Martinez-Mir A, Liu J, Gordon D, Weiner MS, Ahmad W, Fine JD, et al. EB simplex superficialis resulting from a mutation in the type VII collagen gene. J Invest Dermatol 2002;118:547-9.
[Google Scholar]
|
| 19. |
Figueira EC, Crotty A, Challinor CJ, Coroneo MT, Murrell DF. Granulation tissue in the eyelid margin and conjunctiva in junctional epidermolysis bullosa with features of laryngo-onycho-cutaneous syndrome. Clin Experiment Ophthalmol 2007;35:163-6.
[Google Scholar]
|
| 20. |
Kirkham J, Robinson C, Strafford SM, Shore RC, Bonass WA, Brookes SJ, Wright JT. The chemical composition of tooth enamel in junctional epidermolysis bullosa. Arch Oral Biol 2000;45:377-86.
[Google Scholar]
|
| 21. |
Darling TN, Bauer JW, Hintner H, Yancey KB. Generalized atrophic benign epidermolysis bullosa. Adv Dermatol 1997;13:87-119.
[Google Scholar]
|
| 22. |
Puvabanditsin S, Garrow E, Samransamraujkit R, Lopez LA, Lambert WC. Epidermolysis bullosa associated with congenital localized absence of skin, fetal abdominal mass, and pyloric atresia. Pediatr Dermatol 1997;14:359-62.
[Google Scholar]
|
| 23. |
Serrano-Martinez MC, Bagan JV, Silvestre FJ, Viguer MT. Oral lesions in recessive dystrophic epidermolysis bullosa. Oral Dis 2003;9:264-8.
[Google Scholar]
|
| 24. |
Castillo RO, Davies YK, Lin YC, Garcia M, Young H. Management of esophageal strictures in children with recessive dystrophic epidermolysis bullosa. J Pediatr Gastroenterol Nutr 2002;34:535-41.
[Google Scholar]
|
| 25. |
Ayman T, Yerebakan O, Ciftcioglu MA, Alpsoy E. A 13-year-old girl with recessive dystrophic epidermolysis bullosa presenting with squamous cell carcinoma. Pediatr Dermatol 2002;19:436-8.
[Google Scholar]
|
| 26. |
Dharma B, Moss C, McGrath JA, Mellerio JE, Ilchyshyn A. Dominant dystrophic epidermolysis bullosa presenting as familial nail dystrophy. Clin Exp Dermatol 2001;26:93-6.
[Google Scholar]
|
| 27. |
Tosti A, Piraccini BM, Scher RK. Isolated nail dystrophy suggestive of dominant dystrophic epidermolysis bullosa. Pediatr Dermatol 2003;20:456-7.
[Google Scholar]
|
| 28. |
Fassihi H, Diba VC, Wessagowit V, Dopping-Hepenstal PJ, Jones CA, Burrows NP, et al. Transient bullous dermolysis of the newborn in three generations. Br J Dermatol 2005;153:1058-63.
[Google Scholar]
|
| 29. |
Intong LRA, Murell DF. How to take skin biopsies for epidermolysis bullosa. Dermatol Clin 2010;28:197-200, vii.
[Google Scholar]
|
| 30. |
Murrell DF. Collection and transport of specimens for epidermolysis bullosa diagnosis. St. George hospital department of dermatology. Available from: http://www:blisters.org.an/AEBDL.html 2006;Ver 1 [Last accessed on 2010 Jan 20].
[Google Scholar]
|
| 31. |
Pohla-Gulo G, Cepada-Valdes R, Hintner H. Immunofluorescence mapping for the diagnosis of epidermolysis bullosa. Dermatol Clin 2010;28:201-10, vii.
[Google Scholar]
|
| 32. |
Castiglia D, Zambruno G. Molecular testing in epidermolysis bullosa. Dermatol Clin 2010;28:223-9.
[Google Scholar]
|
| 33. |
Pfendner EG, Nakano A, Pulkkinen L, Christiano AM, Uitto J. Prenatal diagnosis for epidermolysis bullosa: A study of 144 consecutive pregnancies at risk. Prenat Diagn 2003;23:447-56.
[Google Scholar]
|
| 34. |
Fassihi H, Eady RA, Mellerio JE, Ashton GH, Dopping-Hepenstal PJ, Denyer JE, et al. Prenatal diagnosis for severe inherited skin disorders: 25 years′ experience. Br J Dermatol 2006;154:106-13.
[Google Scholar]
|
| 35. |
De Benedittis M, Petruzzi M, Favia G, Serpico R. Oro-dental manifestations in Hallopeau-Siemens type recessive dystrophic epidermolysis bullosa. Clin Exp Dermatol 2004;29:128-32.
[Google Scholar]
|
| 36. |
Ingen-Housz-Oro S, Blanchet-Bardon C, Vrillat M, Dubertret L. Vitamin and trace metal levels in recessive dystrophic epidermolysis bullosa. J Eur Acad Dermatol Venereol 2004;18:649-53.
[Google Scholar]
|
| 37. |
Caldwell-Brown D, Stern RS, Lin AN, Carter DM. Lack of efficacy of phenytoin in recessive dystrophic epidermolysis bullosa. N Engl J Med 1992;327:163-7.
[Google Scholar]
|
| 38. |
Hansen SK, Veien NK. Oxytetracycline in epidermolysis bullosa simplex. A double-blind placebo-controlled trial. J Eur Acad Dermatol Venereol 1996;6:277-8.
[Google Scholar]
|
| 39. |
Langan SM,Williams HC. A systematic review of randomized controlled trials of treatments for inherited forms of epidermolysis bullosa. Clin Exp Dermatol 2008;34:20-5.
[Google Scholar]
|
| 40. |
Featherstone C. Epidermolysis Bullosa: From Fundamental Molecular Biology to Clinical Therapies. J Invest Dermatol 2007;127:256-9.
[Google Scholar]
|
| 41. |
Dunnill MG, Rodeck CH, Richards AJ, Atherton D, Lake BD, Petrou M, et al. Use of type VII collagen gene (COL7A1) markers in prenatal diagnosis of recessive dystrophic epidermolysis bullosa. J Med Genet 1995;32:749-50.
[Google Scholar]
|
| 42. |
D′Alessio M, Zambruno G, Charlesworth A, Lacour JP, Meneguzzi G. Immunofluorescence analysis of villous trophoblasts: A tool for prenatal diagnosis of inherited epidermolysis bullosa with pyloric atresia. J Invest Dermatol 2008;128:2815-9.
[Google Scholar]
|
Fulltext Views
11,215
PDF downloads
3,656





![[Table - 1]](#tbl_ijdvl_2011_77_4_431_82393_t1.jpg){kind=link}
![[Table - 2]](#tbl_ijdvl_2011_77_4_431_82393_t2.jpg){kind=link}
![[Figure - 1]](#fig_ijdvl_2011_77_4_431_82393_f3.jpg){kind=link}