Translate this page into:
Generalized Dowling–Degos disease with hypopigmented lesions: A diagnostic challenge
2 Department of Dermatology, Katihar Medical College, Katihar, Bihar, India
3 Department of Dermatology, KPC Medical College and Hospital, Kolkata, West Bengal, India
Correspondence Address:
Piyush Kumar
Department of Dermatology, Katihar Medical College, Katihar, Bihar
India
| How to cite this article: Ghosh A, Kumar P, Das A. Generalized Dowling–Degos disease with hypopigmented lesions: A diagnostic challenge. Indian J Dermatol Venereol Leprol 2018;84:70-72 |
Sir,
Dowling–Degos disease (DDD) is a rare autosomal dominant, progressive, reticulate pigmentary disorder. It usually appears after puberty. It is clinically characterized by hyperpigmented macules, papules and comedone-like lesions on flexures and perioral pitted scars.[1],[2] The pathogenesis of DDD has not been fully elucidated. However, loss of functional mutations in the keratin 5 gene has been described. Follicular pathology has also been attributed to its genesis.[3]
Rarely, hypopigmented macules and papules are found in DDD and are noted in a variant called generalized DDD.[4] Generalized DDD has been uncommonly discussed in the Indian literature. Hence, we report a case of generalized DDD with typical clinical and histopathological findings.
A 23-year-old otherwise healthy gentleman, born of a non consanguineous parentage, presented with multiple asymptomatic hyperpigmented macules and papules on forehead which started to appear 8 years back and slowly progressed to involve the face, neck and trunk [Figure 1a]. In addition, he developed multiple hypopigmented lesions on the lower trunk during the course of the disease [Figure 1b]. Family history was unremarkable. Cutaneous examination revealed multiple hyperpigmented macules and papules over the face, neck and trunk. Lower trunk revealed numerous hypopigmented macules and papules. Hands, feet, mucosae, nails and hair were spared. Routine investigations were within normal limits. A provisional diagnosis of epidermodysplasia verruciformis was made. Dyschromatosis universalis hereditaria and dyschromatosis symmetrica hereditaria were considered as clinical differentials. Skin biopsy was done from both hyperpigmented and hypopigmented lesions. Histopathological examination from both the samples showed circumscribed foci of epidermal proliferation as thin, elongated rete ridges with antler-like branching at places. In addition, biopsy from the hyperpigmented lesion showed increased basal pigmentation as well as increased melanophages in the papillary dermis [Figure 2a]. On the other hand, biopsy from the hypopigmented lesions showed increased pigmentation only at the bottom of rete ridges [Figure 2b]. Absent melanophages and vascular ectasia of papillary dermis were appreciated in hypopigmented lesion. The surprising biopsy findings helped us clinch the diagnosis, and it was established as a case of generalized DDD. Galli Galli disease (GGD) is a rare acantholytic variant of DDD and presents with hyperpigmented lesions in flexures, scaly erythematous plaques, comedo-like lesions and pitted perioral scars. Acantholysis is considered sine qua non for the diagnosis of GGD and differentiates GGD from DDD.[5] We biopsied a total of five lesions (three hyperpigmented and two hypopigmented lesions) and none of them showed acantholysis on histopathology, thereby ruling out the diagnosis of GGD in our case.
 |
| Figure 1a: Numerous hyperpigmented macules and papules on the neck |
 |
| Figure 1b: Numerous hypopigmented macules and papules on the trunk |
 |
| Figure 2a: Histopathology from hyperpigmented lesions showing elongated rete ridges, increased pigmentation at the bottom of rete ridges as well as basilar pigmentation and melanophages (H and E, ×400) |
 |
| Figure 2b: Histopathology from hypopigmented lesions showing thin, elongated rete ridges with increased pigmentation at their tip and vascular ectasia of papillary dermis (H and E, ×400) |
Generalized DDD is a rare variant of DDD and clinically resembles dyschromatosis universalis hereditaria (DUH) and dyschromatosis symmetrica hereditaria (DSH), as summarized in [Table - 1]. Generalized DDD is clinically characterized by hyperpigmented as well as hypopigmented lesions present throughout the body, as seen in our case.[4],[6] Our case did not have comedone-like lesions or perioral pitted scars seen in classical DDD. However, cases described recently by Naveen et al. exhibited open comedones and multiple pitted scars.[7] Both hyperpigmented and hypopigmented lesions show similar and classical histopathological findings of DDD – downward narrow elongation of rete ridges with increased pigmentation, preferentially affecting the tip of these rete ridges. One finding worth mentioning is the presence of diffuse basal pigmentation in hyperpigmented lesions, which is notably absent in hypopigmented lesions.[4],[6] Histopathology in our case echoed similar findings. Moreover, we found hyperpigmented lesions having prominent melanophages, which were minimal or absent in hypopigmented lesions. Hypopigmented lesions showed vascular ectasia of papillary dermis. Further studies are needed to establish or refute these histopathological differences in two types of lesions, as seen in our case. The occurrence of hypopigmented lesions in DDD is intriguing as histopathology features of both hyperpigmented and hypopigmented lesions are quite similar and pathomechanism behind the development of hypopigmented lesions is not understood. Verma et al. described a family of 25 members of DDD/GGD with hypopigmented lesions on trunk and considered that hypopigmented lesions in DDD may be underreported in the Indian population.[8] Rowley et al. proposed that hypopigmented lesions could be a manifestation of subclinical acantholysis,[9] and Verma et al. did find acantholysis in hypopigmented lesions in their cases. However, histopathology from the two hypopigmented lesions we biopsied did not reveal any acantholysis. Rarely, DDD may present with hypopigmented lesions only, adding to the enigma around hypopigmented lesions in DDD.[6]
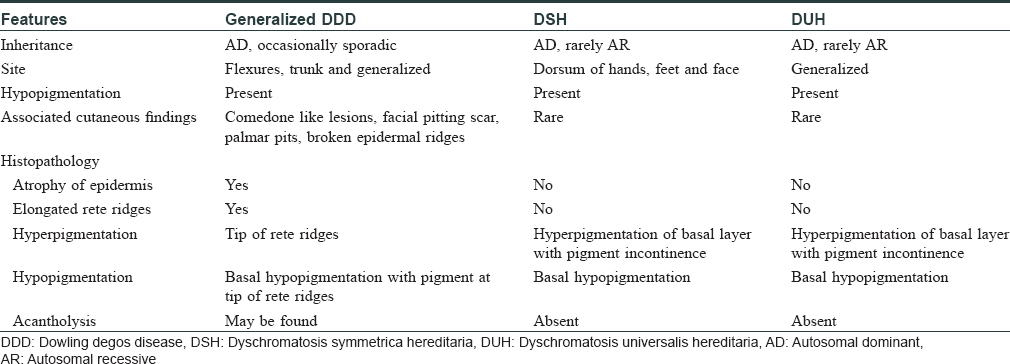
Reticulate pigmentary disorders mimic each other clinically and may have overlapping clinical features, further adding to the diagnostic dilemma. DSH and DUH present with macules only and are characterized by the absence of papules, thereby allowing differentiation from DDD. GGD could be a clinical differential in our case but was ruled out as acantholysis was consistently absent in all samples. Other reticulate pigmentary disorders like dermatopathia pigmentosa reticularis, epidermolysis bullosa with mottled pigmentation and Naegeli-Franceschetti-Jadassohn syndrome are associated with distinct findings on hair, nail, skin, palm and mucosa.[10],[11] Presence or absence of such findings help us in making a clinical diagnosis. Mucocutaneous and systemic examination in our case, apart from the findings mentioned earlier, was unremarkable. Furthermore, histopathological findings in all five lesions we biopsied were diagnostic of DDD.
To conclude, DDD can present with hypopigmented macules and papules along with hyperpigmented lesions (as noted in our case) or rarely, with hypopigmented lesions only. Awareness of different clinical variations is essential as such cases may lack other classical findings of DDD (pitted perioral scars and comedone-like lesions), thereby making clinical diagnosis challenging.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
| 1. |
Sandhu K, Saraswat A, Kanwar AJ. Dowling-Degos disease with dyschromatosis universalis hereditaria-like pigmentation in a family. J Eur Acad Dermatol Venereol 2004;18:702-4.
[Google Scholar]
|
| 2. |
Thami GP, Jaswal R, Kanwar AJ, Radotra BD, Singh IP. Overlap of reticulate acropigmentation of Kitamura, acropigmentation of Dohi and Dowling-Degos disease in four generations. Dermatology 1998;196:350-1.
[Google Scholar]
|
| 3. |
Singh S, Khandpur S, Verma P, Singh M. Follicular Dowling Degos disease: A rare variant of an evolving dermatosis. Indian J Dermatol Venereol Leprol 2013;79:802-4.
[Google Scholar]
|
| 4. |
Wu YH, Lin YC. Generalized Dowling-Degos disease. J Am Acad Dermatol 2007;57:327-34.
[Google Scholar]
|
| 5. |
Desai CA, Virmani N, Sakhiya J, Khopkar U. An uncommon presentation of Galli-Galli disease. Indian J Dermatol Venereol Leprol 2016;82:720-3.
[Google Scholar]
|
| 6. |
Pickup TL, Mutasim DF. Dowling-Degos disease presenting as hypopigmented macules. J Am Acad Dermatol 2011;64:1224-5.
[Google Scholar]
|
| 7. |
Naveen KN, Athaniker SB, Hegde SP, Shetty R, Radha H, Parinitha SS, et al. Atypical cases of Dowling-Degos disease. Indian Dermatol Online J 2016;7:99-102.
[Google Scholar]
|
| 8. |
Verma S, Pasternack SM, Rütten A, Ruzicka T, Betz RC, Hanneken S, et al. The first report of KRT5 mutation underlying acantholytic dowling-degos disease with mottled hypopigmentation in an Indian family. Indian J Dermatol 2014;59:476-80.
[Google Scholar]
|
| 9. |
Rowley MJ, Nesbitt LT Jr., Carrington PR, Espinoza CG. Hypopigmented macules in acantholytic disorders. Int J Dermatol 1995;34:390-2.
[Google Scholar]
|
| 10. |
Adya KA, Inamadar AC, Palit A. Reticulate dermatoses. Indian J Dermatol 2014;59:3-14.
[Google Scholar]
|
| 11. |
Sardana K, Goel K, Chugh S. Reticulate pigmentary disorders. Indian J Dermatol Venereol Leprol 2013;79:17-29.
[Google Scholar]
|
Fulltext Views
7,264
PDF downloads
1,419





![[Table - 1]](#tbl_ijdvl_2018_84_1_70_219241_t5.jpg){kind=link}