Translate this page into:
Identification of GJB6 gene mutation in an Indian man with Clouston syndrome
2 Department of Pediatrics, All India Institute of Medical Sciences, New Delhi, India
Correspondence Address:
Nidheesh Agarwal
Flat No. 001, Fateh Apartment, New Fatehpura, Udaipur, Rajasthan
India
| How to cite this article: Agarwal N, Singh PK, Gupta K, Gupta N, Kabra M. Identification of GJB6 gene mutation in an Indian man with Clouston syndrome. Indian J Dermatol Venereol Leprol 2016;82:697-700 |
Sir,
Clouston syndrome (MIM 129500), also known as hidrotic ectodermal dysplasia, is a rare autosomal dominant genetic disorder characterized by generalized hypotrichosis, dystrophic nails and hyperkeratotic palms and soles. It is particularly common in the French Canadian population of Southwest Quebec. We present an Indian man with typical clinical features of Clouston syndrome who was found to have a known mutation in the GJB6 gene.
A 45-year-old man presented to the dermatology outpatient department of Geetanjali Medical College and Hospital in Udaipur, Rajasthan, with complaints of excessively thickened skin of the palms and soles. The thickened skin restricted movements of his fingers and disrupted his daily functioning. He gave a history of similar features in one of his children. There was no history of similar lesions in any of his siblings, parents or other family members. None of his family members could be examined.
On examination, the patient was found to have severe hyperkeratosis of the palms and soles which led to clawing of the hands [Figure - 1]. Nails were dystrophic in both hands and feet. The fingernails were thickened, overcurved, discolored [Figure - 2] and associated with tufting of the terminal phalanges [Figure - 1]. Further, the patient had thin and sparse scalp hair [Figure - 3]a. Other hair-bearing regions (eyebrows, eyelids, axillae, pubic region) also had very minimal hair [Figure - 3]b. There was no history of consanguinity, hypohidrosis, abnormal dentition and any hearing or visual complaints. A provisional diagnosis of hidrotic ectodermal dysplasia was made.
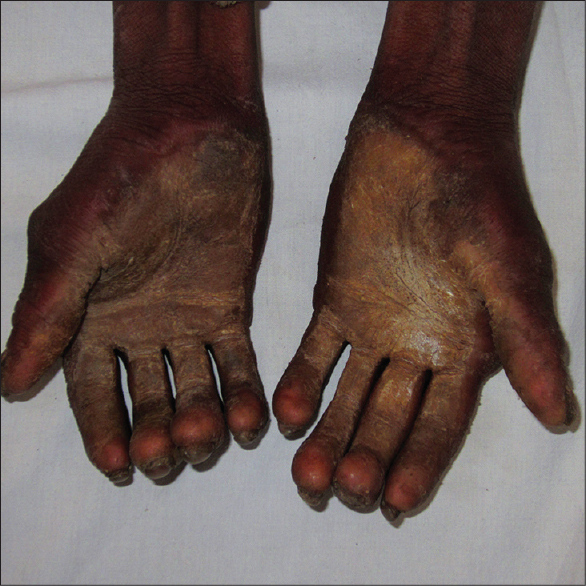 |
| Figure 1: Severe hyperkeratosis of palms leading to clawing, along with bulbous terminal phalanges |
 |
| Figure 2: Thickened, overcurved and discolored finger nails |
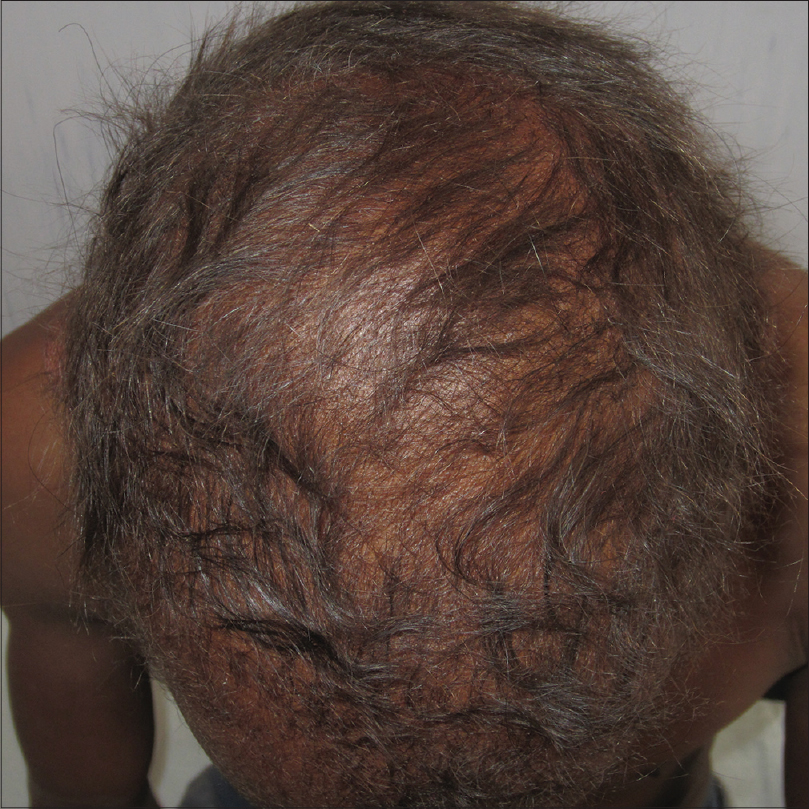 |
| Figure 3: (a) Thin and sparse scalp hair |
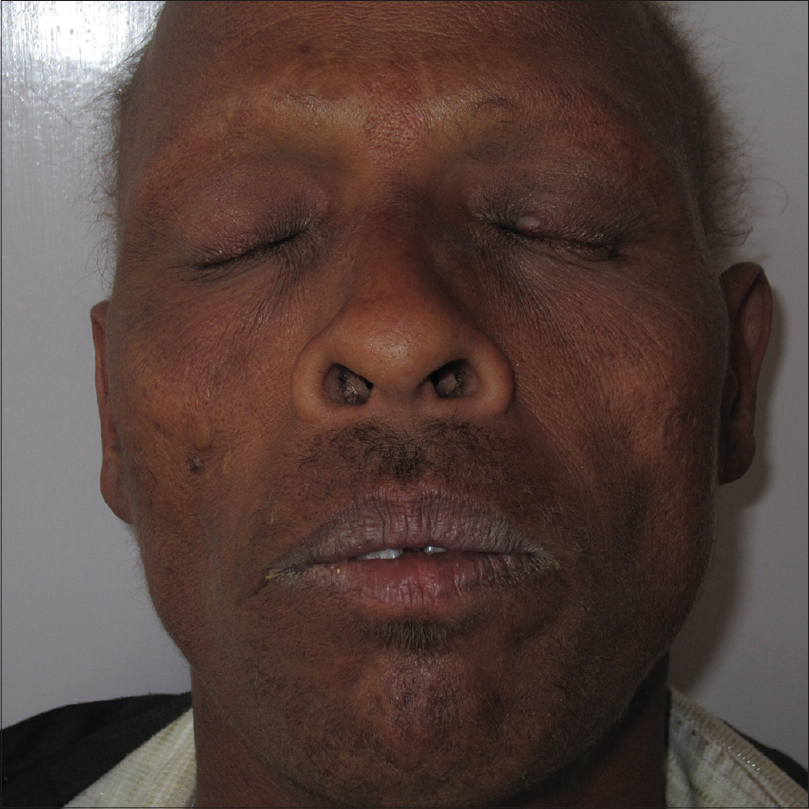 |
| Figure 3: (b) Sparse eyebrows, eyelashes and facial hair |
A peripheral blood sample was collected in an ethylenediaminetetraacetic acid (EDTA) tube and DNA was isolated by the salting out method.[1] Coding exon 3 of the GJB6 gene was amplified by polymerase chain reaction (PCR) and the amplification checked on 2% agarose gel. Primers used are shown in [Table - 1]. The polymerase chain reaction-amplified product was incubated with exonuclease I and shrimp alkaline phosphatase to remove the unincorporated primers and nucleotides. Bidirectional Sanger deoxyribonucleic acid sequencing was done using ABI Prism BigDye Terminator Cycle sequencing ready reaction kit v3.1 (Applied Biosystems, USA), followed by ethanol/ethylenediamine tetraacetic acid/sodium acetate precipitation. The precipitate was dissolved in 10 µL Hi-Di formamide with denaturation and capillary electrophoresis using an ABI 3130 Genetic Analyzer. Sequencing results were analyzed using Chromas Pro software (http://technelysium.com.au) and compared with the reference sequence of GJB6 in the NCBI database (http://www.ncbi.nlm.nih.gov/). A reported heterozygous missense mutation c. 31G>A (p.G11R) was identified, resulting from a glycine-to-arginine amino acid change [Figure - 4].
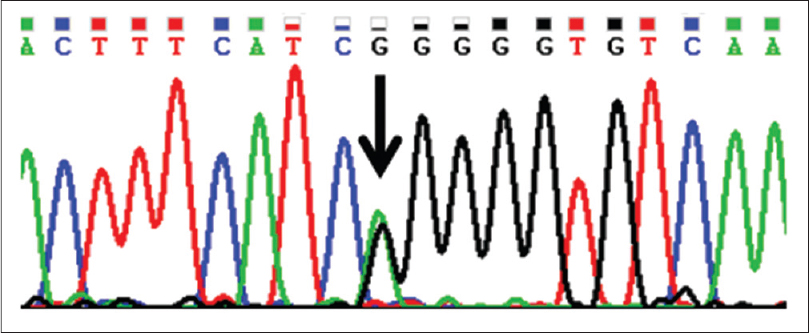 |
| Figure 4: A heterozygous c. 31G>A change in the GJB6 gene |
Clouston syndrome is a rare genetic disease caused by mutations in connexin genes. Connexins or gap junction proteins are a family of structurally related transmembrane proteins which establish direct cell-to-cell communication and are responsible for the movement of molecules and ions across adjacent cells. They may be classified into three major groups (GJA, GJB and GJC) based on sequence homology. Each combination of connexins has different qualities of permeability highly significant in terms of function. Mutations in connexins result in hereditary peripheral neuropathy, complex conotruncal heart malformations, autosomal dominant forms of cataract and hearing loss. Erythrokeratoderma variabilis, keratitis-ichthyosis-deafness syndrome, Vohwinkel syndrome and Clouston syndrome are cutaneous disorders resulting from connexin mutations.
Mutations in GJB6 gene (and in some cases, GJA1 and GJB2 genes) are responsible for Clouston syndrome. To date, five different mutations have been reported in GJB6 gene with phenotypic features of Clouston syndrome. Mutations p.G11R and p.A88V have been described by Lamartine et al. in multiple ethnic populations, p.V37E by Smith et al. in Scottish patients and p.D50N by Baris et al. in Israeli patients.2-4 Recently, Liu et al. have found that the combination of a novel mutation N14S in GJB6 and a mutation F191L in GJB2 played a pathogenic role in Clouston syndrome.[5] Mutations in GJA1 (p.V41L) and an R127H heterozygous variants of GJB2 have also been found responsible for Clouston syndrome.[6] Out of all these, p.G11R is the most commonly reported mutation, seen in the French Canadian population as well as in many other ethnic populations of the world.
We could find only two previous reports of Clouston syndrome in Indians. One was of a large Gujarati family with 41 affected individuals spanning five generations, but genetic mutations were not explored in this family.[7] The other report described a p.A88V mutation detected by Lamartine et al. in a patient of Indian ethnicity.[2]
Our patient presented with typical features, and since he denied any similar features in his parents, it was probably a sporadic case. The mutation detected, c. 31G>A (p.G11R) in the GJB6 gene, is a known mutation.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has given his consent for his images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
| 1. |
Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 1988;16:1215.
[Google Scholar]
|
| 2. |
Lamartine J, Munhoz Essenfelder G, Kibar Z, Lanneluc I, Callouet E, Laoudj D, et al. Mutations in GJB6 cause hidrotic ectodermal dysplasia. Nat Genet 2000;26:142-4.
[Google Scholar]
|
| 3. |
Smith FJ, Morley SM, McLean WH. A novel connexin 30 mutation in Clouston syndrome. J Invest Dermatol 2002;118:530-2.
[Google Scholar]
|
| 4. |
Baris HN, Zlotogorski A, Peretz-Amit G, Doviner V, Shohat M, Reznik-Wolf H, et al. A novel GJB6 missense mutation in hidrotic ectodermal dysplasia 2 (Clouston syndrome) broadens its genotypic basis. Br J Dermatol 2008;159:1373-6.
[Google Scholar]
|
| 5. |
Liu YT, Guo K, Li J, Liu Y, Zeng WH, Geng SM. Novel mutations in GJB6 and GJB2 in Clouston syndrome. Clin Exp Dermatol 2015;40:770-3.
[Google Scholar]
|
| 6. |
Kellermayer R, Keller M, Ratajczak P, Richardson E, Harangi F, Mérei E, et al. Bigenic connexin mutations in a patient with hidrotic ectodermal dysplasia. Eur J Dermatol 2005;15:75-9.
[Google Scholar]
|
| 7. |
Radhakrishna U, Blouin JL, Mehenni H, Mehta TY, Sheth FJ, Sheth JJ, et al. The gene for autosomal dominant hidrotic ectodermal dysplasia (Clouston syndrome) in a large Indian family maps to the 13q11-q12.1 pericentromeric region. Am J Med Genet 1997;71:80-6.
[Google Scholar]
|
Fulltext Views
1,920
PDF downloads
1,036





![[Figure - 1]](#fig_ijdvl_2016_82_6_697_190855_f1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2016_82_6_697_190855_f2.jpg){kind=link}
![[Figure - 3]](#fig_ijdvl_2016_82_6_697_190855_f3.jpg){kind=link}
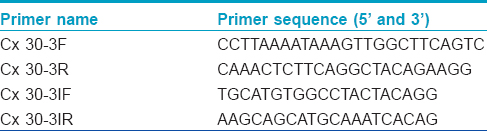
![[Table - 1]](#tbl_ijdvl_2016_82_6_697_190855_t6.jpg){kind=link}
![[Figure - 4]](#fig_ijdvl_2016_82_6_697_190855_f4.jpg){kind=link}