Translate this page into:
Keratoderma-like T cell dyscrasia: A report of 13 cases and its distinction from mycosis fungoides palmaris et plantaris
2 Department of Pathology and Laboratory Medicine, Weill Medical College of Cornell University, New York, New York 10065; Department of Dermatology, University of Colorado Anschutz Medical Campus, Aurora, Colorado 80045, USA
Correspondence Address:
Cynthia M Magro
Department of Pathology and Laboratory Medicine, Weill Medical College of Cornell University, Box 58, Room F-309, 1300 York Avenue, New York, New York 10065
USA
| How to cite this article: Magro CM, Nguyen GH. Keratoderma-like T cell dyscrasia: A report of 13 cases and its distinction from mycosis fungoides palmaris et plantaris. Indian J Dermatol Venereol Leprol 2016;82:395-403 |
Abstract
Background: Atypical epitheliotropic T cell lymphocytic infiltrates are commonly encountered in routine and consultative dermatopathology practices and typically do not represent mycosis fungoides. Other conditions can mimic certain light microscopic and phenotypic findings encountered in mycosis fungoides, comprising a diverse spectrum of conditions including the lymphomatoid drug reaction, collagen vascular disease, viral hypersensitivity reactions and cutaneous T cell dyscrasia. Aims: To examine biopsies obtained from cutaneous T cell dyscrasia localized to the palms and soles and to evaluate whether it exhibits a morphologic and pathogenetic continuum with mycosis fungoides plantaris et palmaris. Methods: We examined 13 biopsies showing an epidermotropic superficial lymphocytic infiltrate from thirteen patients who presented with a palmar and/or plantar keratoderma without other sites of cutaneous involvement. Conventional light microscopy, immunophenotyping and clonality studies were carried out. The clinical features were recorded. Results: Biopsies showed a variably dense, superficial, angiocentric CD4 or CD8 dominant lymphocytic infiltrate accompanied by a non-destructive pattern of epidermotropism. Low-grade cerebriform atypia along with variable diminution in the expression of CD7 and CD62L was noted. In three cases, statins were suspected to be the cause. Due to lack of familiarity with the entity, treatment interventions were inconsistent and not aggressively pursued. There was no evidence of disease progression to mycosis fungoides in any case. Limitations: The limitations of this study include the lack of long-term follow up and information on the nature of the therapeutic interventions and responses to treatment. Conclusion: The spectrum of cutaneous lymphoid dyscrasias should be expanded to include cases manifesting as palmo-plantar keratoderma. These cases are to be distinguished from mycosis fungoides palmaris et plantaris. As with other forms of cutaneous lymphoid dyscrasia, the lesions tend to be persistent. The course however, is indolent in most cases.Introduction
Atypical epitheliotropic T cell lymphocytic infiltrates represent a common reaction pattern encountered in routine and consultative dermatopathology practice and may be caused by a diverse variety of diseases. A subset of these infiltrates is the T cell lymphomas, of which mycosis fungoides is the most common epidermotropic T cell lymphoma. Other epidermotropic T cell lymphomas including Sézary syndrome, adult T cell leukemia lymphoma and primary cutaneous aggressive cytotoxic CD8+ epidermotropic T cell lymphoma may produce a similar pattern of epidermal colonization.[1],[2] Furthermore, there are a number of conditions that can mimic certain aspects of the light microscopic and phenotypic findings encountered in mycosis fungoides, especially in regard to the baseline cerebriform cytomorphology and the pattern of migration into the epidermis.[3] The majority of these infiltrates fall into four categories: cutaneous T cell dyscrasias, lymphomatoid drug reactions, the interface dermatitis of collagen vascular disease and excessive Type IV immune responses to various triggers such as viruses and contactants.[1] The cutaneous T cell dyscrasias represent indolent cutaneous clonal T cell proliferative disorders manifesting a low risk of disease progression to mycosis fungoides. Unlike the reactive lymphomatoid conditions such as those related to collagen vascular disease, a well-defined trigger does not exist.[4],[5] While there are certain pathogenetic commonalities with mycosis fungoides, the cutaneous dyscrasias warrant recognition as a unique form of hematologic dyscrasia distinct and separate from mycosis fungoides as the vast majority of patients have non-progressive disease confined to the skin.[5]
Cutaneous T cell dyscrasias demonstrate low-grade lymphoid atypia and phenotypic abnormalities of lymphocytes similar to those encountered in mycosis fungoides including a loss in the expression of certain pan T cell markers, namely CD7 and CD62L.[6] Based on prior studies, the greatest diminution in expression among pan T cell markers is with CD62L. CD7 can also be decreased although not to the magnitude seen with CD62L.[1] Clonality has been described in the cutaneous T cell dyscrasias using a highly sensitive methodology that involves a gene scanning software for fragment size analysis and quantitative measurement of fluorescence intensity.[7],[8] Using a less sensitive technique for the detection of T cell clones, polyclonality is more characteristic. These divergent findings may reflect the relatively small percentage of clonally restricted T cells that are presumably causative of the condition.[9] In lesions of cutaneous lymphoid dyscrasias, much of the infiltrate may be reactive, possibly directed at the aberrant T cell clone and may contribute toward the tendency of these lesions to spontaneously regress.[10]
The more commonly recognized T cell dyscrasias are alopecia mucinosa, pityriasis lichenoides, pigmented purpuric dermatosis and large plaque parapsoriasis. The full spectrum of cutaneous lesions falling under the rubric of cutaneous lymphoid dyscrasia also encompasses folliculotropic T cell lymphocytosis, atypical lymphocytic lobular panniculitis and syringolymphoid hyperplasia with alopecia.[1] In these entities, excluding atypical lymphocytic lobular panniculitis, the established and published histologic criteria in recognizing the cutaneous lymphoid dyscrasias include cerebriform lymphoid atypia and an intraepidermal architectural disposition of lymphocytes similar to mycosis fungoides but lacking sufficient cytologic atypia to warrant categorization as mycosis fungoides. Loss of CD7 can be similar to that in mycosis fungoides but other phenotypic abnormalities such as a significant loss of CD5 or a double negative phenotype in the atypical intraepidermal lymphoid cells is unusual. We describe a novel form of epitheliotropic T cell dyscrasia characterized by a persistent palmar and plantar keratoderma. We propose the designation of keratoderma-like epitheliotropic T cell dyscrasia.
Methods
By using a natural-language search, we uncovered 13 cases of keratoderma-like T cell dyscrasia from the Weill Cornell (New York, NY, USA) dermatopathology database during the period January 1, 2006, to July 31, 2013. All cases were reviewed and interpreted by CMM. Skin biopsy specimens in all cases had been submitted for diagnostic assessment of a persistent keratoderma. In each case, routine light microscopic analysis and phenotypic and molecular studies were conducted on paraffin-embedded, formalin-fixed tissue. The comprehensive phenotypic panel included antibodies to β-F1, CD2, CD3, CD4, CD5, CD7, CD62L and CD8. A standard T-cell receptor γ gene rearrangement assay was performed. These immunohistochemical and molecular methods have been previously reported.[9] This study received institutional review board approval under protocol number 0710009479 from Weill Medical College of Cornell University, New York, USA.
Results
Clinical summary
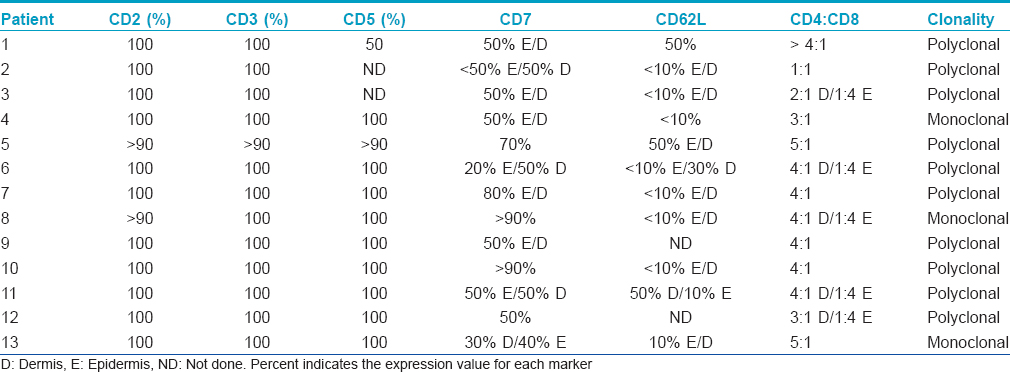
This case series included a total of thirteen patients exhibiting a similar presentation comprising scaly erythematous plaques on the palms and/or soles [Table - 1]. Nail changes were seen in one patient with an established history of psoriasis.
There were nine women and four men. The patients' ages ranged from 9 years to 78 years (mean age of 56). A detailed account of the drug history and clinical features are provided in [Table - 1]. In all cases, only the palms and/or soles were involved which showed localized or diffuse erythema and scaliness. In most patients, the keratoderma followed a persistent and/or waxing and waning course over a few years. The most common clinical diagnoses were dyshidrotic eczema and psoriasis. The remainder of the clinical exam was unremarkable. Ten of the patients were taking a statin, antidepressant, antihistamine, estrogen, calcium channel blocker and/or angiotensin converting enzyme inhibitor. A statin was the most commonly used drug. In three cases, discontinuing the statin was associated with resolution of the eruption [Table - 1], patients 6, 7 and 10]. In one case, the eruption occurred shortly after starting the statin; however, the patient only discontinued the statin briefly and continues on the statin with persistent disease [Table - 1], number 10]. Various treatment modalities were used including topical steroids in most, keratolytic agents in one (patient 2), anti-tumor necrosis factor alpha therapy in one (patient 3), methotrexate in one (patient 4), drug cessation in four (patients 6–8, and 10), azathioprine in one (number 8), intramuscular triamcinolone in one (number 11), narrow band ultraviolet B (patient 13) and oral psoralen and ultraviolet A (patient 3). The patient receiving an anti-tumor necrosis factor for presumed psoriasis did not experience any improvement (patient 3), while the patient receiving methotrexate initially had resolution of the keratoderma (patient 4). Topical steroids provided minimal relief and, in one patient, use for a few years led to steroid atrophy with persistent disease (patient 2). Narrow band ultraviolet B therapy was recommended in two patients; however, one patient did not return for treatment (patient 1) and another showed some improvement but had persistent disease (patient13). Four patients have experienced regression of keratoderma: two who discontinued their statin (patients 6 and 7), one patient who received intramuscular steroids (patient 11) and one patient treated with methotrexate (patient 4). Two typical cases showing classic palmar and plantar scaly erythema are illustrated in [Figure - 1]. The disease did not extend to other skin areas and features of mycosis fungoides did not develop in any patient.
 |
| Figure 1: Patient 4 had a persistent scaly eruption of the (a) hands and (b) feet for 2 years which was resistant to topical steroids. The patient was on albuterol, hydroxyurea, loratadine, and aspirin. |
Light microscopic findings
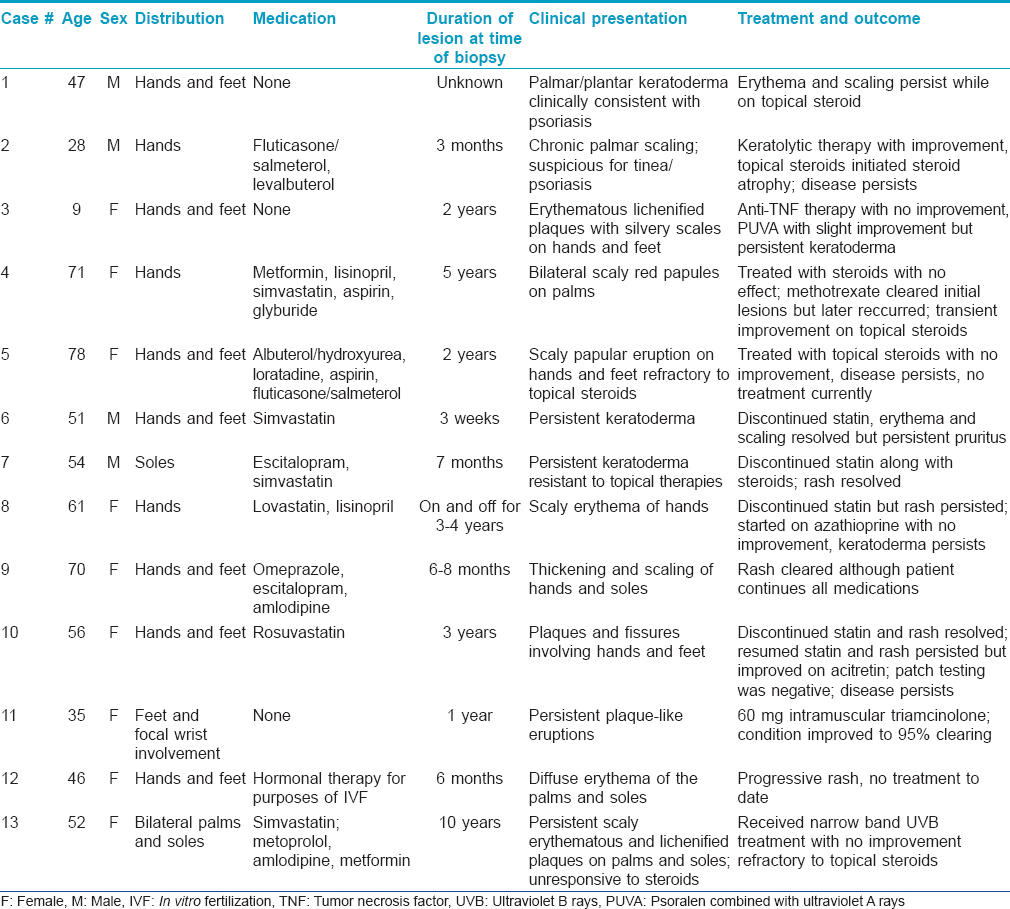
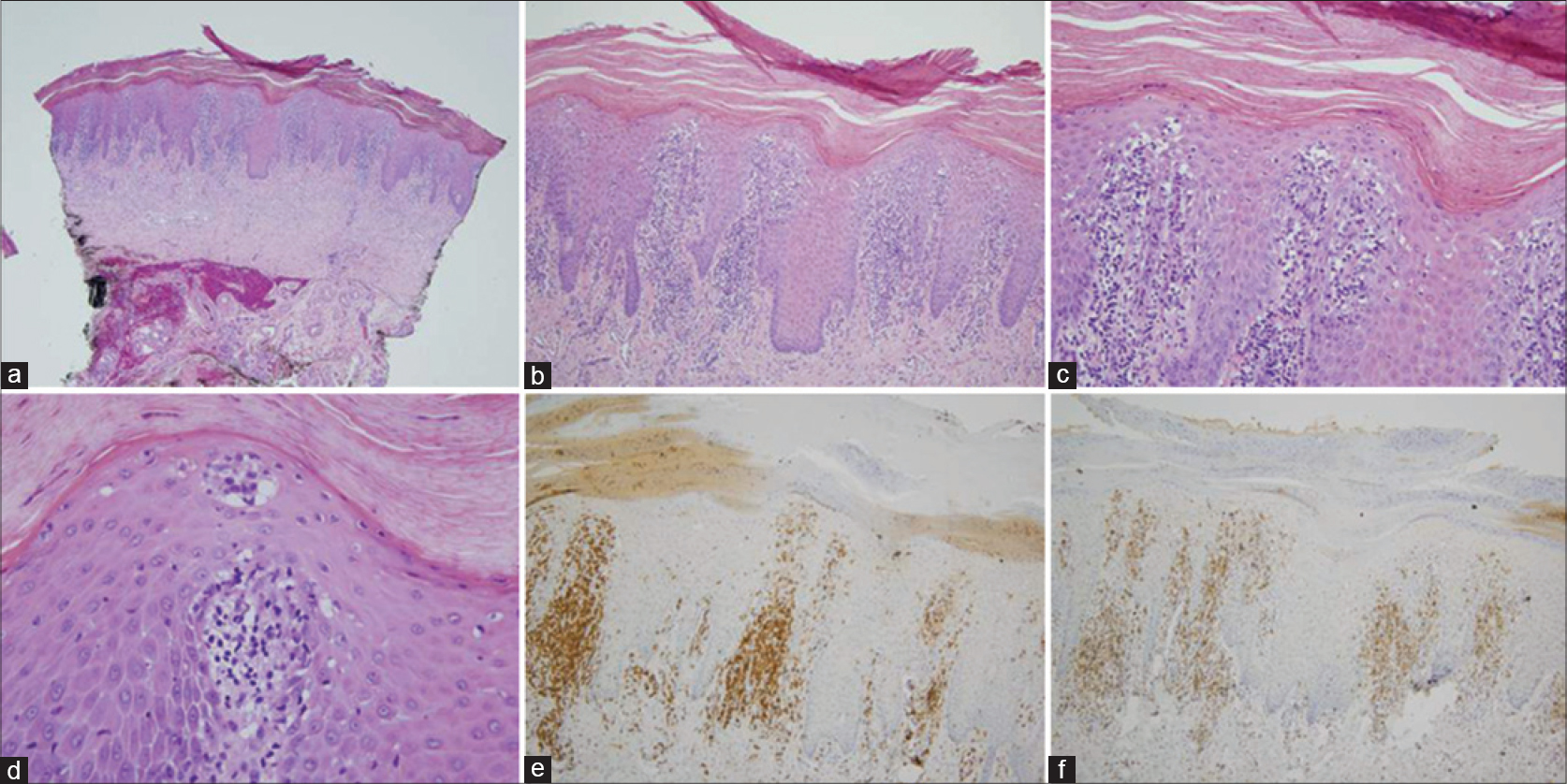
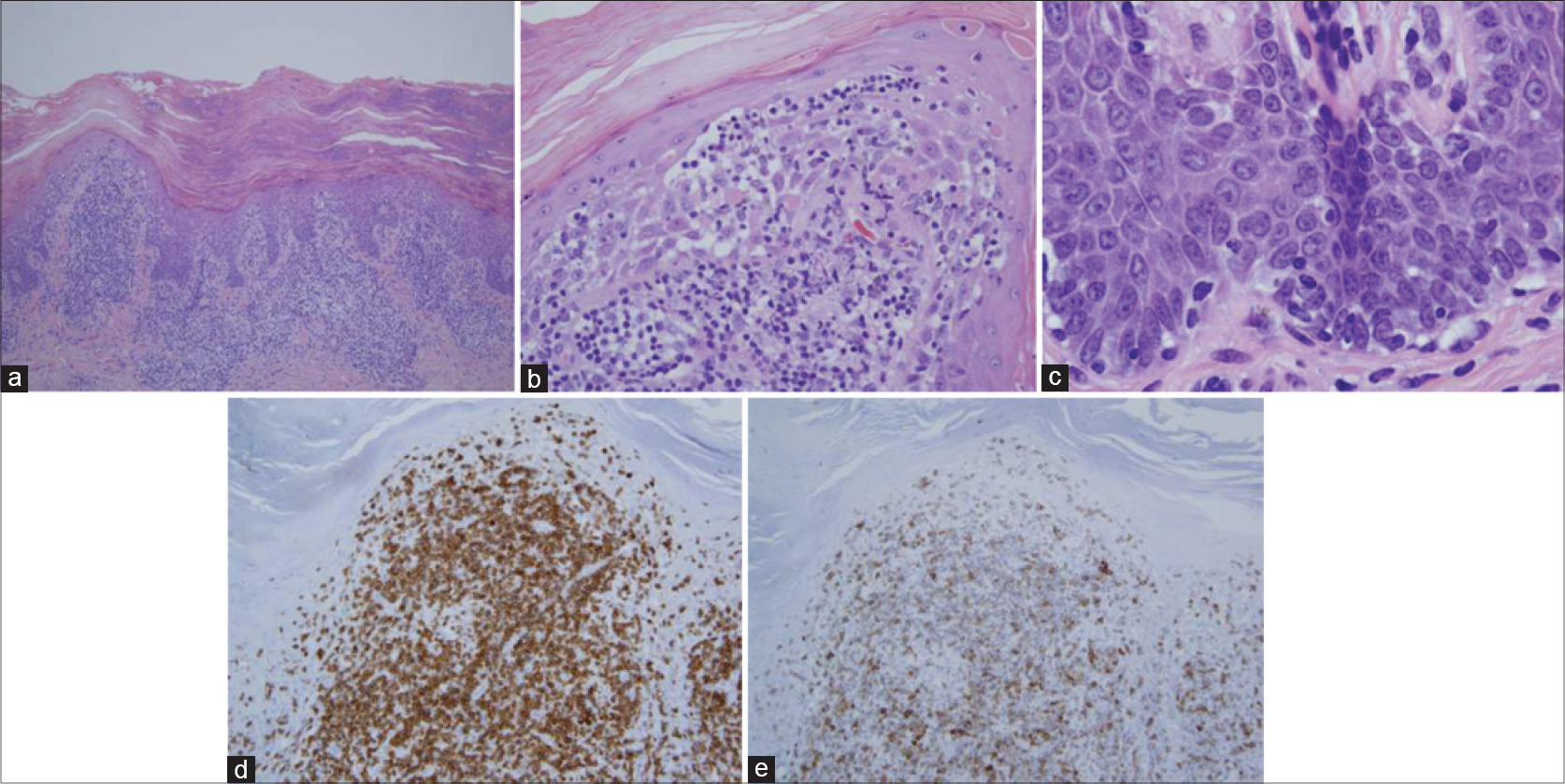
All thirteen cases had a very similar appearance. There was a psoriasiform epidermal hyperplasia of moderate to marked degree with prominent hyperkeratosis (corresponding clinically to keratoderma) [Figure - 2]a, [Figure - 3]a and [Figure - 4]a. In the majority of cases, the pattern of keratinization was one of orthohyperkeratosis with minimal parakeratosis [Figure - 2]b and [Figure - 3]b. More extensive parakeratosis was also seen in some cases. The dermal lymphocytic infiltrate was confined to the superficial dermis in all cases. The infiltrate was perivascular with accentuation around the superficial vascular plexus including the capillaries in the dermal papillae [Figure - 2]a and [Figure - 2]b, [Figure - 3]a and [Figure - 3]b and [Figure - 4]b. Conspicuous red cell extravasation was observed in half of the cases without concomitant mural and/or luminal fibrin deposition. Foci of prominent suprabasilar exocytosis into the mid and upper spinous layer were a very characteristic feature observed in 11 of the 13 cases [Figure - 2]b, [Figure - 2]c, [Figure - 2]d, [Figure - 3]b and [Figure - 4]b. There was infiltration of the basal layer by atypical lymphocytes in each case [Figure - 3]c and [Figure - 4]c; subtle vacuolar change was noted in 6 cases. However, in all cases there were areas where the lymphocytes assumed a passive pattern of migration into the epidermis [Figure - 3]c. In particular, the lymphocytes were present in the epidermis in a quiescent fashion without evoking a significant epithelial response. Even when there was dyskeratosis, it was largely unaccompanied by lymphocyte satellitosis and involved the upper spinous layers in 10 of the 13 cases. All cases showed some lymphocytes with cerebriform nuclei; however, the greater degree of nuclear contour irregularity typical of mycosis fungoides was not seen [Figure - 2]d, [Figure - 3]c and [Figure - 4]d. There was a dearth of other inflammatory cells apart from histiocytes [Figure - 2]d. These findings were histologically most reminiscent of pityriasis lichenoides chronica although orthohyperkeratosis was commonly noted rather than the dominant parakeratotic pattern seen in pityriasis lichenoides chronica.
 |
| Figure 2: (a) The biopsy of patient 5 shows a prominent psoriasiform pattern of epidermal hyperplasia with striking hyperkeratosis. There is a supervening superficial lymphocytic infiltrate which is angiocentric and epitheliotropic (H and E, ×4). (b and c) The pattern of keratinization is primarily orthohyperkeratosis with minimal parakeratosis. The granular cell layer is diminished. There is colonization of the basal layer by lymphocytes accompanied by small aggregates of lymphocytes within the mid spinous layer of the epidermis. The morphology captured in this biopsy is reminiscent of pityriasis lichenoides (H and E, ×10 and ×20) (d) The cohesive aggregate of lymphocytes and histiocytes in the upper spinous layer of the epidermis mimics a Pautrier's microabscess but is different by virtue of the lack of greater lymphoid atypia and the number of admixed histiocytes (H and E, ×40). (e) The extent of the lymphocytic infiltrate is highlighted by the CD3 stain (×20). (f) There is a minor reduction in the expression of CD7 (×20) (c and d) Patient 7 had erythema and scaling of the feet for 7 months which was resistant to steroids. The patient was on escitalopram and simvastatin |
 |
| Figure 3: The biopsy of patient 12 is remarkably similar to an earlier case. (a) There is a prominent pattern of epidermal hyperplasia with striking hyperkeratosis. There is both orthohyperkeratosis and parakeratosis. (H and E, ×4). (b) A prominent superficial perivascular lymphocytic infiltrate is identified. There is migration of lymphocytes into the epidermis with extension into the upper spinous layer of the epidermis (H and E, ×20). (c) Foci of passive colonization of the basal layer by atypical lymphocytes are noted. (H and E, ×40). (d) The lymphocytes within the epidermis stain positively for CD3. (×20). (e) In contrast, there is a significant reduction in the extent of immunoreactivity for CD7. (H and E, ×20). |
 |
| Figure 4: (a) The biopsy of patient 6 shows a mild psoriasiform pattern of epidermal hyperplasia. The epidermis is surmounted by a thick orthohyperkeratotic scale. There is a supervening perivascular lymphocytic infiltrate primarily involving the superficial dermis. Focal migration of lymphocytes into the epidermis is observed. Note that the pattern of superficial dermal infiltration differs from that seen in mycosis fungoides by virtue of the lack of true band-like lymphocytic infiltration. (H and E, ×4) (b) There is migration of lymphocytes into the epidermis with preferential involvement of the acrosyringium. (H and E, ×10). (c) In this photomicrograph, there is a passive pattern of lymphocyte migration into the epidermis, recapitulating a pattern seen in mycosis fungoides (H and E, ×20). (d). Higher power magnification shows mild nuclear contour irregularity including cells with a cerebriform appearance (H and E, ×40). The eruption was temporally associated with ingestion of a statin and resolved completely upon discontinuing the statin |
Immunophenotypic profile
The infiltrates were composed almost exclusively of T cells which were highlighted by the pan T cell markers CD2, CD3 and CD5 [Figure - 2]e and [Figure - 3]d. A reduction of CD7 was observed in 11 of the 13 cases and ranged from 20% to 50% amidst intraepidermal lymphocytes and from 30% to 70% in the dermal component [Figure - 2]f and [Figure - 3]e and [Table - 2]. In 11 of the 13 cases for which specimens were available for CD62L staining, there was a reduction in the expression of CD62L of about 90% in 8 of the 11 tested cases. In all cases, CD4 lymphocytes predominated over CD8 lymphocytes in the dermis. However, there was a reversal of the CD4:CD8 ratio in lymphocytes within the epidermis in 5 cases. A summary of the phenotypic profile is presented in [Table - 2].
Molecular studies
Clonality detection was carried out for all samples as previously described.[9] Polyclonality was detected in the skin samples of 10 of 13 patients. In three cases, a monoclonal result was observed.
Discussion
We have described a series of patients who presented with a cutaneous eruption localized to the palms and soles. In each case, the eruption was clinically described as a keratoderma. However, the histopathological features were those of an atypical epidermotropic T cell proliferation, resulting in a diagnostic conundrum. The findings of low-grade cerebriform lymphoid atypia, the pattern of passive basilar colonization and the aberrant phenotypic profile characterized by a reduction in the expression of the pan T cell markers CD7 and CD62L led us to a histopathological diagnosis of epidermotropic T cell dyscrasia. Intraepidermal lymphocytes were largely unaccompanied by any evidence of epithelial injury directly attributable to lymphocyte-keratinocyte interaction. In contradistinction, the classic pattern of lymphocyte satellitosis around degenerating keratinocytes typifies immunologically mediated forms of interface dermatitis such as erythema multiforme and graft versus host disease. The histology had many features in common with pityriasis lichenoides chronica, a classic and well established form of cutaneous T cell dyscrasia, although the clinical features were not compatible with that diagnosis.
On light microscopy, the biopsies showed a fairly reproducible morphology exhibiting many overlapping features with pityriasis lichenoides. Hyperkeratosis was marked and typically dominated by an orthohyperkeratotic pattern. The infiltrate in the superficial dermis was primarily angiocentric as opposed to band-like, although there was prominent migration of lymphocytes into the epidermis. A basal layer pattern of colonization mimicking mycosis fungoides was noted but tended to be more focal. There were small cohesive aggregates of lymphocytes involving the more superficial layers of the epidermis along with spinous layer dyskeratosis. The greater extent of reduction of CD62L as compared to CD7 is a finding previously reported in other T cell dyscrasias.[8] In contrast, the extent of diminution of CD7 and CD62L in mycosis fungoides is characteristically marked for both markers and similar in degree. In 5 of the 13 cases, the atypical intraepidermal lymphocyte was of the CD8 subset which was somewhat similar to findings seen in pityriasis lichenoides and the hypopigmented interface variant of cutaneous T cell dyscrasia.[11],[12] All three of these T cell dyscrasias are associated with dyskeratosis and vacuolar interface change, alterations which could in part be triggered by the cytotoxic properties of the infiltrating CD8+ lymphocytes.[1]
Keratoderma is a clinical term for a persistent erythematous scaly dermatosis localized to the palms and soles.[13] The process may reflect a disorder of keratinization such as hereditary keratoderma of Marie Unna Thoste and keratoderma climactericum.[14],[15] The majority of the keratodermas are inflammatory conditions of varied etiologies comprising psoriasis, dishydrotic eczema, secondary syphilis, and keratodermia blenorrhagicum associated with Reiter's disease. A nutritional dermatosis, specifically pellagra and a paraneoplastic form of keratoderma (i.e., Bazex syndrome), are uncommon forms of keratoderma.[16],[17] Only a minority of cases are attributable to a T cell dyscrasia, primarily in the context of Sézary syndrome and mycosis fungoides. We were unable to find any previous reports of a keratoderma-like T cell dyscrasia not representing T cell lymphoma.
In half of the cases presented in this series, the process was presumed to be triggered by immune dysregulatory drug therapy, most frequently statins. In three cases, the eruption resolved quickly after cessation of the implicated agent. One could use the designation of drug-associated reversible T cell dyscrasia to describe this form of lymphomatoid drug reaction that closely mimics endogenous forms of T cell dyscrasia.[18],[19] The other cases were felt to represent endogenous forms of T cell dyscrasia that tended to have a protracted course often resistant to topical steroids.
The differential diagnosis for this form of T cell dyscrasia includes mycosis fungoides palmaris et plantaris, a variant of mycosis fungoides limited to the palms and the soles.[20] We found published reports of 26 cases of the latter, described in [Table - 3].[20],[21],[22],[23],[24],[25],[26],[27],[28],[29],[30] It was first described by Resnik et al. in 1995 as a rare variant comprising only 0.6% of mycosis fungoides cases.[20] Similar to keratoderma-like T cell dyscrasia, lesions of mycosis fungoides palmaris et plantaris exhibit marked hyperkeratosis, a passive pattern of epidermal colonization and cerebriform lymphoid atypia including basilar epidermotropism.[23] Given the striking similarity to keratoderma-like T cell dyscrasia, including an indolent clinical course in most cases, it is possible that some cases reported as mycosis fungoides palmaris et plantaris represent T cell dyscrasia, especially since involvement of the palms and soles is infrequent in conventional mycosis fungoides (excluding Sezary syndrome). Unlike keratoderma-like T cell dyscrasia which typically shows a superficial perivascular lymphocytic infiltrate as the dominant pattern of dermal infiltration, lesions of mycosis fungoides palmaris et plantaris demonstrate a lichenoid pattern and are accompanied by laminated dermal fibroplasia and markedly hyperconvoluted lymphocytes. Plasma cells and eosinophils are noted in the infiltrate in mycosis fungoides while they are not seen in the keratoderma-like T cell dyscrasias. Pautrier's microabscesses, characteristic for mycosis fungoides palmaris et plantaris, are not seen in keratoderma-like T cell dyscrasia. On the other hand, keratoderma-like T cell dyscrasia shows collections of Langerhans cells and lymphocytes localized to the superficial layers of the epidermis, identical to those seen in pityriasis lichenoides. This differs from the classic Pautrier's microabscess that is present lower in the epidermis in a parabasilar location and is composed almost exclusively of cerebriform lymphocytes without any interposed Langerhans cells. T-cell receptor γ gene rearrangement analysis shows clonality in mycosis fungoides palmaris et plantaris, a finding identified in only 3 of our 13 cases.[7]

The limitation of this study is the lack of long-term follow-up. Many patients were lost to follow-up and/or did not return for further treatments. Due to the unfamiliarity of the referring clinician with the diagnosis, there was no uniform treatment regimen that was successfully executed in a number of cases. A more regimented approach to treatment with serial follow-up assessment would be ideal for future cases. Once the entity of keratoderma T cell dyscrasia becomes a more recognized one analogous to large plaque parapsoriasis, pityriasis lichenoides and pigmented purpuric dermatosis, a better defined therapeutic approach will hopefully emerge. Untreated, the eruption can be quite persistent.
Conclusion
We propose that keratoderma-like T cell dyscrasia is a clinically and pathologically distinct entity separate from mycosis fungoides palmaris et plantaris. Like other forms of T cell dyscrasia, the clinical course appears to be indolent without any documentation of disease progression to mycosis fungoides. The lack of familiarity with the entity resulted in a clinical approach that was varied, ranging from keratolytic agents to the use of methotrexate. Further studies are needed to determine the optimal treatment for this recalcitrant dermatitis.
Acknowledgement
We would like to thank Natalie Drucker for her editorial assistance.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
| 1. |
Magro CM, Crowson AN, Mihm M. The Cutaneous Lymphoid Proliferations: A Comprehensive Textbook of Lymphocytic Infiltrates of the Skin. Hoboken, NJ: John Wiley and Sons; 2007. p. 267-88.
[Google Scholar]
|
| 2. |
Willemze R, Jaffe ES, Burg G, Cerroni L, Berti E, Swerdlow SH, et al. WHO-EORTC classification for cutaneous lymphomas. Blood 2005;105:3768-85.
[Google Scholar]
|
| 3. |
Nashan D, Faulhaber D, St, Stb S, Luger TA, Stadler R. Mycosis fungoides: A dermatological masquerader. Br J Dermatol 2007;156:1-10.
[Google Scholar]
|
| 4. |
Magro CM, Crowson AN. Folliculotropic T-cell lymphocytosis as a distinct form of pilotropic T-cell dyscrasia. Am J Clin Pathol 2011;135:221-9.
[Google Scholar]
|
| 5. |
Guitart J, Magro C. Cutaneous T-cell lymphoid dyscrasia: A unifying term for idiopathic chronic dermatoses with persistent T-cell clones. Arch Dermatol 2007;143:921-32.
[Google Scholar]
|
| 6. |
Magro CM, Sachdeva MP, Crowson AN, Barusevicius A, Baran PN, Kovatich AJ. The application of a monoclonal antibody to CD62L on paraffin-embedded tissue samples in the assessment of the cutaneous T-cell infiltrates. J Cutan Pathol 2005;32:12-20.
[Google Scholar]
|
| 7. |
Plaza JA, Morrison C, Magro CM. Assessment of TCR-beta clonality in a diverse group of cutaneous T-Cell infiltrates. J Cutan Pathol 2008;35:358-65.
[Google Scholar]
|
| 8. |
Chen M, Deng A, Crowson AN, Srinivasan M, Yearsley KH, Jewell S, et al. Assessment of T-cell clonality via T-cell receptor-gamma rearrangements in cutaneous T-cell-dominant infiltrates using polymerase chain reaction and single-stranded DNA conformational polymorphism assay. Appl Immunohistochem Mol Morphol 2004;12:373-9.
[Google Scholar]
|
| 9. |
Magro CM, Schaefer JT, Crowson AN, Li J, Morrison C. Pigmented purpuric dermatosis: Classification by phenotypic and molecular profiles. Am J Clin Pathol 2007;128:218-29.
[Google Scholar]
|
| 10. |
Magro C, Crowson AN, Kovatich A, Burns F. Pityriasis lichenoides: A clonal T-cell lymphoproliferative disorder. Hum Pathol 2002;33:788-95.
[Google Scholar]
|
| 11. |
Magro CM, Crowson AN, Morrison C, Li J. Pityriasis lichenoides chronica: Stratification by molecular and phenotypic profile. Hum Pathol 2007;38:479-90.
[Google Scholar]
|
| 12. |
Wenzel J, Genzel J, I, Distelmaier M, Uerlich M, Mikus S, Bieber T, et al. The role of cytotoxic skin-homing CD8+ lymphocytes in cutaneous cytotoxic T-cell lymphoma and pityriasis lichenoides. J Am Acad Dermatol 2005;53:422-7.
[Google Scholar]
|
| 13. |
Klaus S, Weinstein GD, Frost P. Localized epidermolytic hyperkeratosis. A form of keratoderma of the palms and soles. Arch Dermatol 1970;101:272-5.
[Google Scholar]
|
| 14. |
K5.1:2 W, Becker A. Indication for the identity of palmoplantar keratoderma type Unna-Thost with type Vwith. Thost's family revisited 110 years later. Acta Derm Venereol 1992;72:120-2.
[Google Scholar]
|
| 15. |
Deschamps P, Leroy D, Pedailles S, Mandard JC. Keratoderma climactericum (Haxthausen's disease): Clinical signs, laboratory findings and etretinate treatment in 10 patients. Dermatologica 1986;172:258-62.
[Google Scholar]
|
| 16. |
DesGroseilliers JP, Shiffman NJ. Pellagra. Can Med Assoc J 1976;115:768-70.
[Google Scholar]
|
| 17. |
Sarkar B, Knecht R, Sarkar C, Weidauer H. Bazex syndrome (acrokeratosis paraneoplastica). Eur Arch Otorhinolaryngol 1998;255:205-10.
[Google Scholar]
|
| 18. |
Magro CM, Crowson AN. Drug-induced immune dysregulation as a cause of atypical cutaneous lymphoid infiltrates: A hypothesis. Hum Pathol 1996;27:125-32.
[Google Scholar]
|
| 19. |
Magro CM, Cruz-Inigo AE, Votava H, Jacobs M, Wolfe D, Crowson AN. Drug-associated reversible granulomatous T cell dyscrasia: A distinct subset of the interstitial granulomatous drug reaction. J Cutan Pathol 2010;37 Suppl 1:96-111.
[Google Scholar]
|
| 20. |
Resnik KS, Kantor GR, Lessin SR, Kadin ME, Chooback L, Cooper HS, et al. Mycosis fungoides palmaris et plantaris. Arch Dermatol 1995;131:1052-6.
[Google Scholar]
|
| 21. |
Goldberg DJ, Stampien TM, Schwartz RA. Mycosis fungoides palmaris et plantaris: Successful treatment with the carbon dioxide laser. Br J Dermatol 1997;136:617-9.
[Google Scholar]
|
| 22. |
Sandwich JT, Davis LS. Mycosis fungoides palmaris et plantaris. Arch Dermatol 1996;132:971.
[Google Scholar]
|
| 23. |
McNiff JM, Schechner JS, Crotty PL, Glusac EJ. Mycosis fungoides palmaris et plantaris or acral pagetoid reticulosis? Am J Dermatopathol 1998;20:271-5.
[Google Scholar]
|
| 24. |
Spieth K, Grundmann-Kollmann M, Runne U, Staib G, Fellbaum C, Wolter M, et al. Mycosis-fungoides-type cutaneous T cell lymphoma of the hands and soles: A variant causing delay in diagnosis and adequate treatment of patients with palmoplantar eczema. Dermatology 2002;205:239-44.
[Google Scholar]
|
| 25. |
Toritsugi M, Satoh T, Higuchi T, Yokozeki H, Nishioka K. A vesiculopustular variant of mycosis fungoides palmaris et plantaris masquerading as palmoplantar pustulosis with nail involvement. J Am Acad Dermatol 2004;51:139-41.
[Google Scholar]
|
| 26. |
Kim ST, Jeon YS, Sim HJ, Kim SH, Kim YK, Suh KS, et al. Clinicopathologic features and T-cell receptor gene rearrangement findings of mycosis fungoides palmaris et plantaris. J Am Acad Dermatol 2006;54:466-71.
[Google Scholar]
|
| 27. |
Topf S, LS, L M, Neisius U, Brabletz T, Simon M Jr, Schuler G, et al. Mycosis fungoides palmaris et plantaris lann unusual variant of cutaneous T-cell lymphoma. Eur J Dermatol 2006;16:84-6.
[Google Scholar]
|
| 28. |
Lambert TJ, Prieto VG, Duvic M. Multiple plaques on the hands and feet. Mycosis fungoides (MF) palmaris et plantaris (MFPP). Arch Dermatol 2007;143:109-14.
[Google Scholar]
|
| 29. |
Jin SP, Jeon YK, Cho KH, Chung JH. Excimer laser therapy (308 nm) for mycosis fungoides palmaris et plantaris: A skin-directed and anatomically feasible treatment. Br J Dermatol 2010;163:651-3.
[Google Scholar]
|
| 30. |
Nakai N, Hagura A, Yamazato S, Katoh N. Mycosis fungoides palmaris et plantaris successfully treated with radiotherapy: Case report and mini-review of the published work. J Dermatol 2014;41:63-7.
[Google Scholar]
|
Fulltext Views
4,895
PDF downloads
2,743





![[Table - 1]](#tbl_ijdvl_2016_82_4_395_181502_t5.jpg){kind=link}
![[Figure - 1]](#fig_ijdvl_2016_82_4_395_181502_f1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2016_82_4_395_181502_f2.jpg){kind=link}
![[Figure - 3]](#fig_ijdvl_2016_82_4_395_181502_f3.jpg){kind=link}
![[Figure - 4]](#fig_ijdvl_2016_82_4_395_181502_f4.jpg){kind=link}
![[Table - 2]](#tbl_ijdvl_2016_82_4_395_181502_t6.jpg){kind=link}
![[Table - 3]](#tbl_ijdvl_2016_82_4_395_181502_t7.jpg){kind=link}