Translate this page into:
Primary benign hypergammaglobulinemic purpura of waldenstrom masquerading as disseminated schamberg's purpura
Correspondence Address:
Balkrishna P Nikam
Krishna Institute of Medical sciences, University, Karad. Dist-Satara, Maharashtra
India
| How to cite this article: Nikam BP, Singh NJ, Shetty DD. Primary benign hypergammaglobulinemic purpura of waldenstrom masquerading as disseminated schamberg's purpura. Indian J Dermatol Venereol Leprol 2011;77:205-208 |
Sir,
Hypergammaglobulinemic purpura of Waldenstrom (HGPW) is characterized by hypergammaglobulinemia, recurring purpura particularly on lower extremities, leukopenia, anemia, an elevated erythrocyte sedimentation rate (ESR), and the presence of rheumatoid factor (RF) indicative of circulating immune complexes. [1],[2] HGPW can be primary or secondary. It is considered primary when there is no other associated disease. Secondary HGPW has a significant association with autoimmune diseases, especially Sjogrens syndrome and lupus erythematosus. [2],[3] We report a case of primary benign HGPW, which presented with recurrent purpuric rash on extremities for the last 10 years and had undergone repeatedly extensive investigations for the same without conclusion.
A 35-year-old female presented with asymptomatic reddish rash on extremities, abdomen and buttocks, recurring for the last 10 years. The rash was most profuse on the lower extremities and aggravated by prolonged standing, walking, traveling and wearing stockings. On cutaneous examination, she was found to have diffuse macules of reddish purpura coalescing densely at lower leg, ankle region and healing with brownish pigmentation as seen in Schamberg′s purpura [[Figure - 1]a]. Purpuric rash was also distributed with decreased density on knees, thighs, buttocks, lower abdomen and flexor aspect of forearm in a reticulated manner [[Figure - 1]b and c]. Over the past 10 years, the patient had consulted many physicians and dermatologists and had undergone a battery of investigations including repeated hemogram (which revealed that she was anemic, had persistent leukopenia and raised ESR) and so was treated with hematinics and occasional blood transfusion. Other investigations like thyroid function tests, liver function tests, renal function tests, urine routine and microscopic examination, chest X-ray, Doppler study of lower limbs, computerized tomography (CT) abdomen and pelvis revealed no abnormality. Tuberculosis enzyme-linked immunosorbent assay (TB ELISA) IgM was positive in 2004 when she was put on AKT for 6 months. (However, primary tuberculosis focus was not detected.) This was with no effect following which she consulted a hematologist who performed investigations which revealed normal coagulation profile and platelet count; anti-phospholipid antibody (APLA) and lupus anticoagulant were negative; Anti nuclear antibody (ANA) was (1:16) borderline positive; and serology for HIV, HBsAg and HCV was negative. Her bone marrow examination was within normal limits. She was treated with iron and folic acid supplements and short courses of oral steroids with no effect. She also consulted many dermatologists who diagnosed her as disseminated Schamberg′s purpura and treated with oral vitamin C, pentoxyphylline and topical steroids without much benefit in treating the recurring rash.
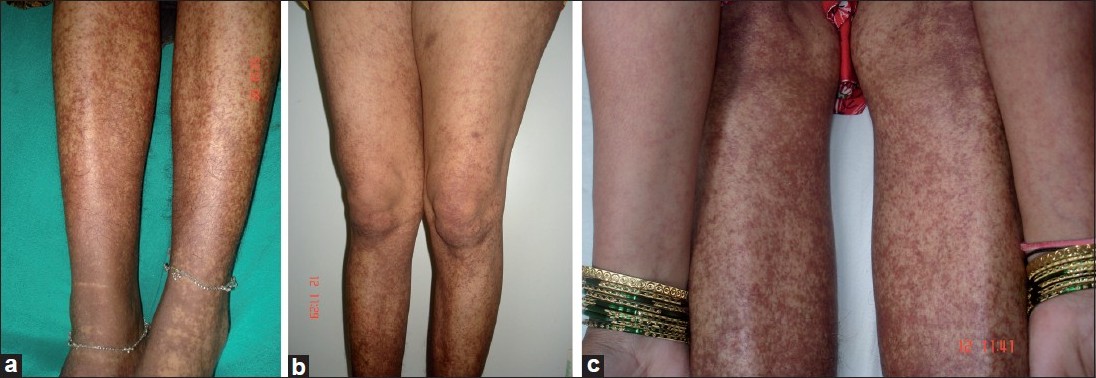 |
| Figure 1: Dense coalescent reddish purpura on (a) lower legs, reticulate purpura on (b) thighs and (c) forearms |
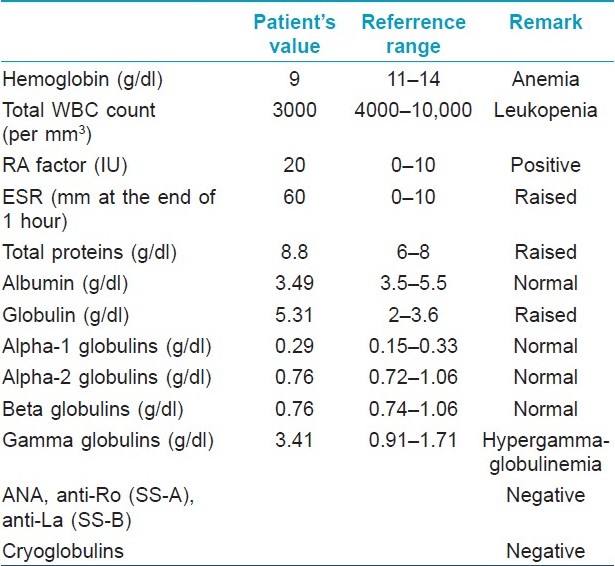
In view of her positive findings in the investigations done in past and clinically recurring benign purpura for many years, the differential diagnosis of HGPW was considered and further investigations were carried out. Skin biopsy from purpuric lesion showed dilated superficial capillaries, extravasation of red blood cells, and sparse mononuclear infiltrate without evident vasculitis, suggestive of noninflammatory purpura [Figure - 2]. Immunofluorescence histological studies were not done. Repeat hemogram findings were consistent with the previous investigations done. Serum protein electrophoresis revealed polyclonal hypergammaglobulinemia [Table - 1]. Although she was not having any history suggestive of connective tissue disease (like joint pain, epilepsy, photosensitivity, etc.) or Sjogren syndrome (like dryness of mouth, eyes, dyspareunia), ANA was repeated and was negative. Anti-Ro (SS-A) and anti-La (SS-B) were also negative. Cryoglobulins were negative.
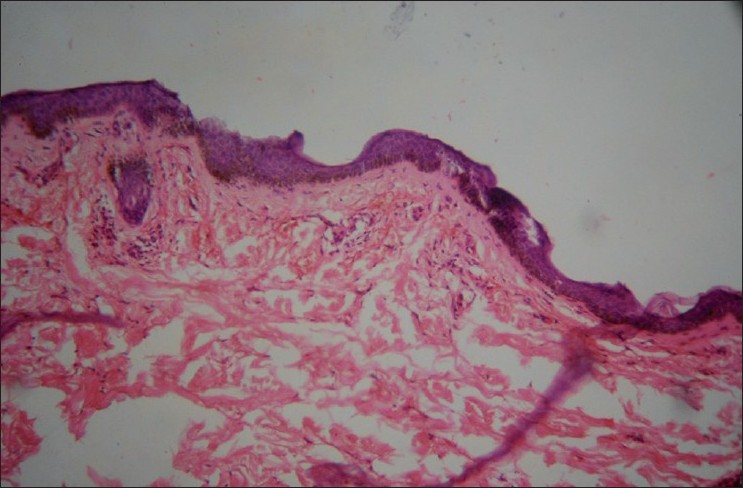 |
| Figure 2: Histopathology showing dilated superficial capillaries, extravasation of red blood cells, and sparse mononuclear infiltrate without evident vasculitis (H and E, ×40) |
Thus, in view of her persistent anemia, leukopenia, raised ESR, positive RA factor and benign polyclonal hypergammaglobulinemia and negative ANA, SS-A, SS-B, the patient was diagnosed as having primary benign HGPW. She was treated for the same with tablet hydroxychloroquine 200 mg twice daily and tablet indomethacin 25 mg once daily. After six months, the rash over the forearm, abdomen and buttocks resolved completely. The rash over the lower legs had partially responded to treatment, with persistent brownish pigmentation [Figure - 3].
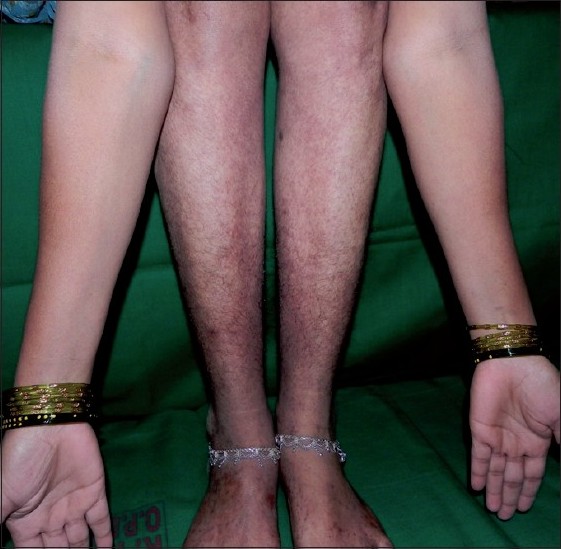 |
| Figure 3: Partial improvement after 6 months of oral hydroxychloroquine. Redness resolved with persistent brownish pigmentation of hemosiderin |
Waldenstrom, in 1952, described three cases of long-standing purpura due to benign long-standing increase in serum globulins. (Two of his patients had sicca symptoms and one had sarcoidosis). [1] The course of the disease is essentially benign but chronic. It usually affects women. Clinically, the pattern of purpura is often non-specific, usually consisting of crops of small erythematous macular or palpable purpuric spots on the lower leg. [2] Reticulate purpura, as seen in our case, has also been described. [4] The lesions may be fleeting. Prolonged walking, standing or sitting with the legs dependant or other reasons of increase in venous pressure (like tight fitting garments, constrictive garters, stockings, heat application) are obvious provocative factors. [2],[3] Some patients have palpable purpura with histological evidence of vasculitis, which has led many authors to classify HGPW as a subset of cutaneous small vessel vasculitis. [3] Histology may or may not show vasculitis; direct immunofluroscence may show immunoglobulin in blood vessel walls and it is suggested that this disorder is an immune complex vasculitis. [4],[5]
As it is an uncommon cause of non-thrombocytopenic purpura, presence of anemia, leukopenia and sometimes prominent immunological abnormalities may lead the clinician to extensive and often unnecessary investigations. [2] The primary form of HGPW is a benign systemic immunoinflammatory disease that requires neither extensive investigations nor any aggressive form of therapy. [3] Lesions usually resolve in about a week but may become confluent and permanent. Indomethacin and hydroxychloroquine may be of value in the treatment of milder disease. [5],[6] In secondary cases, the treatment is aimed at the underlying disease and includes steroids, cyclophosphamide, mycophenolate mofetil, interferon-alpha, rituximab or plasmapheresis. Rare deaths are related to the associated cryoglobulin disease.
The case is reported with the objective of increasing awareness on HGPW and with the aim of increasing the frequency of its diagnosis, especially in the patient group with non thrombocytopenic purpura. Evaluation of serum proteins along with electrophoresis in cases of nonthombocytopenic purpura may lead to early diagnosis of HGPW, avoiding unnecessary burden of multiple misdiagnosis, futile investigations and the psychological strain of the disease.
| 1. |
Waldenstrom J. Three new cases of purpura hyperglobulinaemica. A study of a long-standing benign increase in serum globulin. Acta Med Scand 1952;266:931-46.
[Google Scholar]
|
| 2. |
Malaviya AN, Kaushik P, Budhiraja S, al-Mutairi M, Nampoory MR, Hussein A, et al. Hypergammaglobulinemic purpura of Waldenström: Report of 3 cases with a short review. Clin Exp Rheumatol 2000;18:518-22.
[Google Scholar]
|
| 3. |
Finder KA, McCollough ML, Dixon SL, Majka AJ, Jaremko W. Hypergammaglobulinemic purpura of Waldenström. J Am Acad Dermatol 1990;23:669-76.
[Google Scholar]
|
| 4. |
Tan E, Ng SK, Tan SH, Wong GC. Hypergammaglonulinaemic purpura presenting as reticulate purpura. Cli Exp Dermatol 1999;24:469-72.
[Google Scholar]
|
| 5. |
Lopez LR, Schocket AL, carr RI, Kohler PF. Lymphocytotoxic antibodies and intermediate immune complexes in hypergammaglobulinaemic purpura of Waldenstrom. Ann Allergy 1988;61:93-6.
[Google Scholar]
|
| 6. |
Senecal JL, Chartier S, Rothfield N. Hypergammaglobulinaemic purpura in systemic autoimmune rheumatic disease: Predictive value of anti-Ro (SSA) and anti-La (SSB) antibodies and treatment with indomethacin and hydroxychloroquine. J Rheumatol 1995;22:868-75.
[Google Scholar]
|
Fulltext Views
6,159
PDF downloads
1,978





![[Figure - 1]](#fig_ijdvl_2011_77_2_205_77470_f2.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2011_77_2_205_77470_f3.jpg){kind=link}
![[Table - 1]](#tbl_ijdvl_2011_77_2_205_77470_t1.jpg){kind=link}
![[Figure - 3]](#fig_ijdvl_2011_77_2_205_77470_f4.jpg){kind=link}