Translate this page into:
Sclerodermoid plaques: A riddle of 'H'
Correspondence Address:
Kinjal D Rambhia
Department of Dermatology, Seth GS Medical College and KEM Hospital, Parel, Mumbai, Maharashtra
India
| How to cite this article: Rambhia KD, Dongre AM, Khopkar US. Sclerodermoid plaques: A riddle of 'H'. Indian J Dermatol Venereol Leprol 2015;81:327 |
Sir,
The H syndrome is an autosomal recessive genodermatosis with cutaneous phenotypes of varying severity and multi-system involvement. Patients suffering from this disorder can be easily mistaken for morphea and connective tissue disorders.
A 12-year-old boy presented with dark colored patches on both lower extremities and trunk since the past 10 years. His other complaints were low height and breathlessness on exertion. Cutaneous examination revealed large hyperpigmented, hypertrichotic, indurated, ichthyotic plaques extending symmetrically from the lower extremities to the mid-truncal area characteristically sparing the knees [Figure - 1] and the medial aspect of buttocks [Figure - 2]. A prominent constriction band with distension of the abdomen and visible veins over the thoraco-abdominal areas were noticed. Examination also revealed hypertelorism, dysmorphic facies, short stature, hypospadias, micropenis, scrotal swelling, accessory nipple, flexion contractures of the knees, and metatarsophalangeal joints. General examination revealed pallor, pedal edema, and inguinal lymphadenopathy.
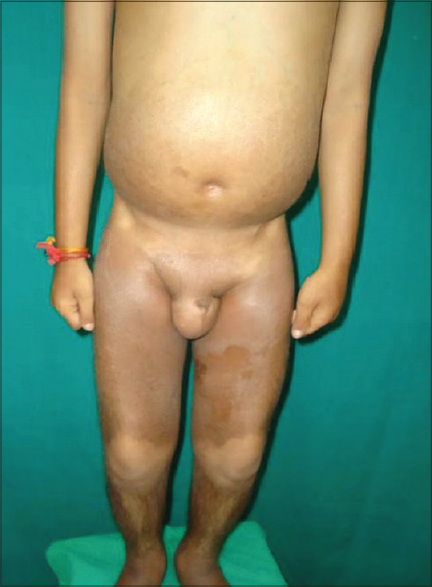 |
| Figure 1: Hyperpigmented, hypertrichotic plaques on lower extremities |
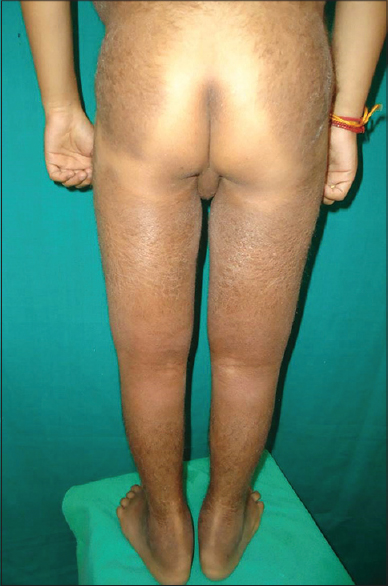 |
| Figure 2: Sclerodermoid plaques sparing the medial parts of buttocks |
Skin biopsy revealed marked fibrosis of the dermis [Figure - 3]a and subcutaneous tissue with diffuse infiltration of histiocytes, intermingled with bundles of dermal collagen [Figure - 3]b. Septal panniculitis with plasma cell infiltration was noted.
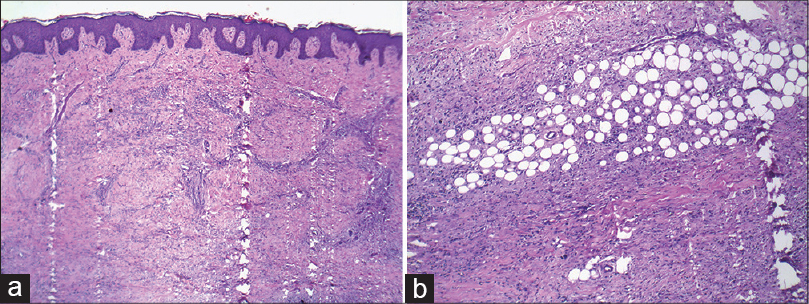 |
| Figure 3: (a) Fibrosis and perivascular infiltrate in the dermis (Hematoxylin and Eosin, ×400). (b) Fibrosis with chronic inflammatory infiltrate (Hematoxylin and Eosin, ×400) |
His hemoglobin was 7.5 g/dl, total leukocytes 7800 cells/mm 3 , and platelet count was 578,000 cells/mm 3 . Erythrocyte sedimentation rate was 132 mm/hr. Liver, renal, thyroid function tests and blood sugar levels were normal. There was hypoalbuminemia (2.2 g/dl), hyperglobulinemia (4.7 g/dl) and hypocalcemia (7.8 mg/dl). Serum viral markers for human immunodeficiency virus (HIV), hepatitis B and C were negative. Antinuclear antibody and anti-dsDNA antibodies were absent. Basal growth hormone (GH) levels were normal. Post-clonidine growth hormone level at 60 min and insulin-like growth factor-1 were reduced. His serum ferritin, reticulocyte count, hemoglobin electrophoresis, serum protein electrophoresis, prothrombin time, and activated partial thromboplastin time were normal. Bone marrow biopsy showed myeloid hyperplasia. Echocardiography revealed atrial septal defect (secundum) and tricuspid regurgitation. Ultrasonography of the abdomen showed mild hepatomegaly.
Hamada and Banka [1] observed hyperpigmented induration over the inner aspects of both thighs, extending to the abdomen in Arab siblings without any systemic involvement and diagnosed the condition as plasma cell panniculitis due to the predominance of plasma cells on histopathology. Marina and Broshtilova described the association of similar cutaneous features with type 1 diabetes mellitus and growth retardation. Prendiville et al. described four patients having similar findings as ′pigmented hypertrichotic dermatosis′. Similar cases were reported as morphea profunda, plasma cell panniculitis and POEMS (polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy and skin changes) syndrome in literature.
The name H syndrome was first coined by Molho-Pessach et al.[2] after reviewing 10 Arab patients with hyperpigmented, hypertrichotic, indurated, ichthyotic plaques on lower extremities with characteristic sparing of knees, scrotal swelling, micropenis, short stature, hepatomegaly and cardiac anomalies as observed in our case. However, hearing loss, dilated scleral vessels, proptosis, hyperglycemia, gynaecomastia, and camptodactyly have also been described, which were absent in our patient. Considering the characteristic cutaneous phenotype, the histological resemblance and the similarity of some associated systemic manifestations of this patient with the cases described by Molho-Pessach, a diagnosis of ′H syndrome′ was made. Genetic testing was not possible due to resource constraints.
Patients of Indian origin described in literature had hypertrichotic, hyperpigmented plaques on lower extremities, short stature, proptosis, gynacomastia, [3] one patient presenting with acute myocardial ischemia due to coronary atherosclerosis. [4]
H syndrome is an autosomal-recessive genodermatosis with multisystem involvement and diverse cutaneous phenotypes. The mean age of onset is 10 years and a male preponderance is observed. Twenty mutations have been identified in the SLC29A3 gene, which encodes human equilibrative nucleoside transporter (hENT3) on the sixth exon. [5] This gene is widely expressed in various organs and is probably a regulator of the inflammatory cascade. A defect in this protein causes disordered immune regulation with development of inflammatory infiltrates in the skin and internal organs. Failure to suppress the inflammation finally results in fibrosis. [6] Further insight into the pathogenesis and the role of hENT3 may lead to a better understanding of the pleiotropic nature of this disease.
| 1. |
Hamadah IR, Banka N. Autosomal recessive plasma cell panniculitis with morphea-like clinical manifestation. J Am Acad Dermatol 2006;54 (Suppl):S189-91.
[Google Scholar]
|
| 2. |
Molho Pessach V, Agha Z, Aamar S, Glaser B, Doviner V, Hiller N, et al. The H syndrome: A genodermatosis characterized by indurated, hyperpigmented, and hypertrichotic skin with systemic manifestations. J Am Acad Dermatol 2008;59:79-85.
[Google Scholar]
|
| 3. |
Priya TP, Philip N, Molho-Pessach V, Busa T, Dalal A, Zlotogorski A. H syndrome: Novel and recurrent mutations in SLC29A3. Br J Dermatol 2010;162:1132-4.
[Google Scholar]
|
| 4. |
Shankarappa RK, Ananthakrishna R, Math RS, Yalagudri SD, Karur S, Dwasrakaprasad R, et al. Accelerated coronary atherosclerosis and H syndrome. BMJ Case Rep 2011;2011: Pii: Bcr0320114019.
[Google Scholar]
|
| 5. |
Molho-Pessach V, Ramot Y, Camilee F, Doviner V, Babay S, Luis SJ, et al. H syndrome: The first 79 patients. J Am Acad Dermatol 2014;70:80-8.
[Google Scholar]
|
| 6. |
Molho-Pessach V, Lerer I, Abeliovich D, Agha Z, Libdeh AA, Broshtilova V, et al. The H syndrome is caused by mutations in the nucleoside transporter hENT3. Am J Hum Genet 2008;83:529-34.
[Google Scholar]
|
Fulltext Views
2,285
PDF downloads
2,738





![[Figure - 1]](#fig_ijdvl_2015_81_3_327_152748_f1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2015_81_3_327_152748_f2.jpg){kind=link}
![[Figure - 3]](#fig_ijdvl_2015_81_3_327_152748_f3.jpg){kind=link}