Translate this page into:
A rare case of phakomatosis pigmentokeratotica associated with unilateral renal hypoplasia
Correspondence Address:
Biju Vasudevan
Department of Dermatology, Base Hospital, Lucknow, Uttar Pradesh
India
| How to cite this article: Sinha A, Kumar V, Verma R, Vasudevan B. A rare case of phakomatosis pigmentokeratotica associated with unilateral renal hypoplasia. Indian J Dermatol Venereol Leprol 2020;86:545-549 |
Sir,
Phakomatosis pigmentokeratotica is a rare and distinct variant of epidermal nevus syndromes which was first described by Happle et al., and characterized by the coexistence of an epidermal nevus (usually nevus sebaceus) and a speckled lentiginous nevus associated with various neurological, skeletal or other extracutaneous defects.[1] Phakomatosis pigmentokeratotica is caused by a postzygotic Harvey rat sarcoma mutation in a multipotent progenitor cell. In this new hypothesis, the respective mutation in the multiple progenitor cells gives rise to both cutaneous and extracutaneous abnormalities noted in phakomatosis pigmentokeratotica.[2] We, herein, report a case of phakomatosis pigmentokeratotica with cutaneous manifestations, in association with rare extracutaneous abnormalities, in the form of unilateral renal hypoplasia and skeletal abnormalities.
A 12-year-old child presented to the dermatology outpatient department, with complaints of multiple dark-colored lesions over face, neck, chest and scalp, present since birth. The lesions over the face, neck and back were relatively flat at birth, but gradually became more verrucous with increasing age. He was born to a nonconsanguineous marriage, through a normal vaginal term delivery, with uneventful antenatal and birth history. There was no history of congenital defects or similar lesions in other family members. He had normal psychomotor development for age.
On examination, multiple dark brown, hyperkeratotic, verrucous plaques following Blaschko's lines were present over the right side of the face [Figure - 1], neck and upper interscapular region, extending to the scalp, leading to localized alopecia on the left temporal area, postauricular region and neck [Figure - 2]. There was a large light brown patch extending unilaterally from the left scapula at the back, left side of the neck and left chest wall in front, extending up to the umbilicus and also covering a portion of the left arm, with multiple small dark brown macules and papules overlying it [Figure - 3] and [Figure - 4]. Thus, clinical examination suggested the existence of two different types of nevi, that is, nevus sebaceus along Blaschko's lines and speckled lentiginous nevus on the left side of the trunk. The orthopedic evaluation showed limb length discrepancy between the two lower limbs and clinically apparent scoliosis [Figure - 5].
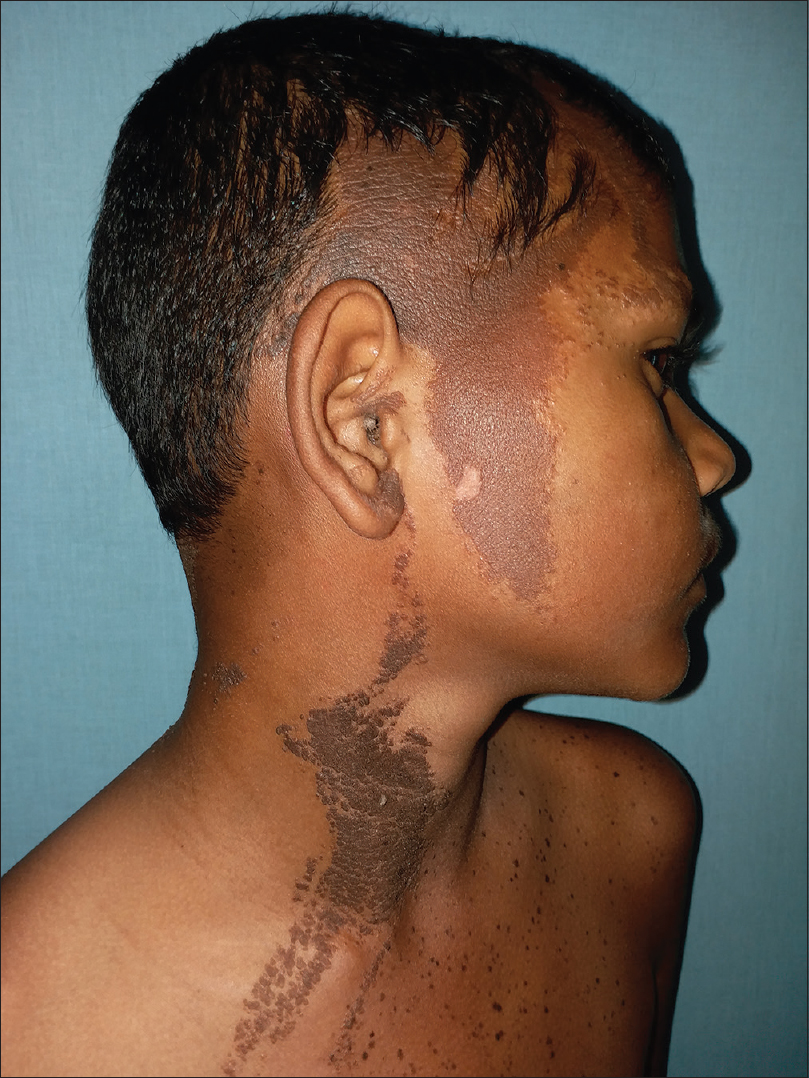 |
| Figure 1: Hyperkeratotic verrucous plaques following Blaschko's lines on the right side of the face and neck |
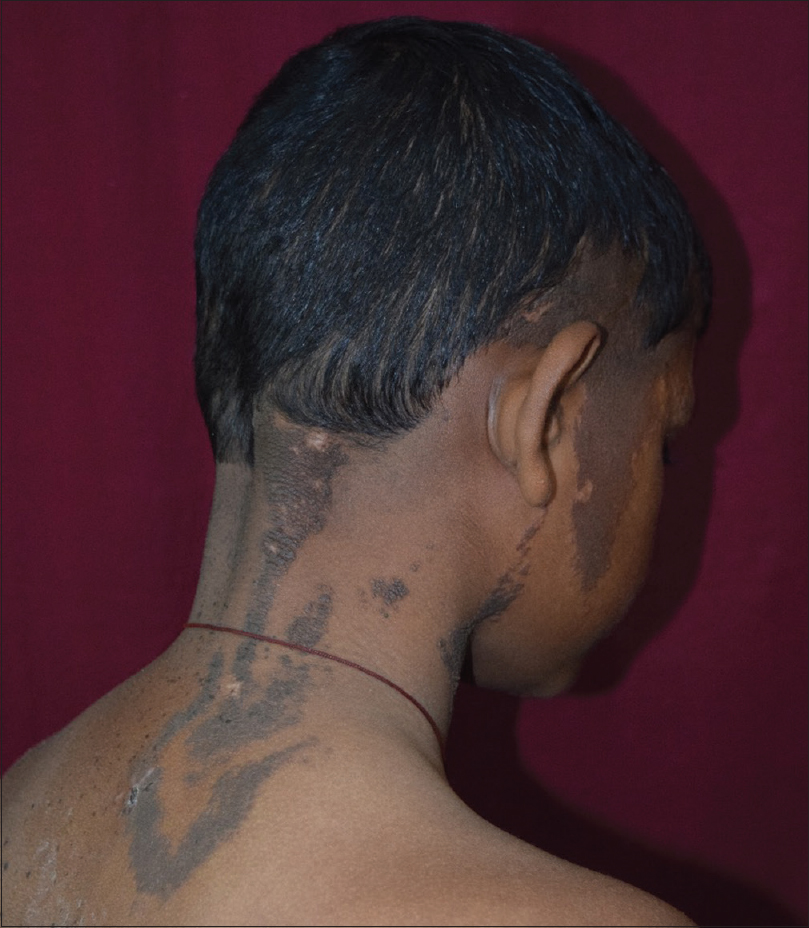 |
| Figure 2: Verrucous plaques over the postauricular region, nape of neck and upper back |
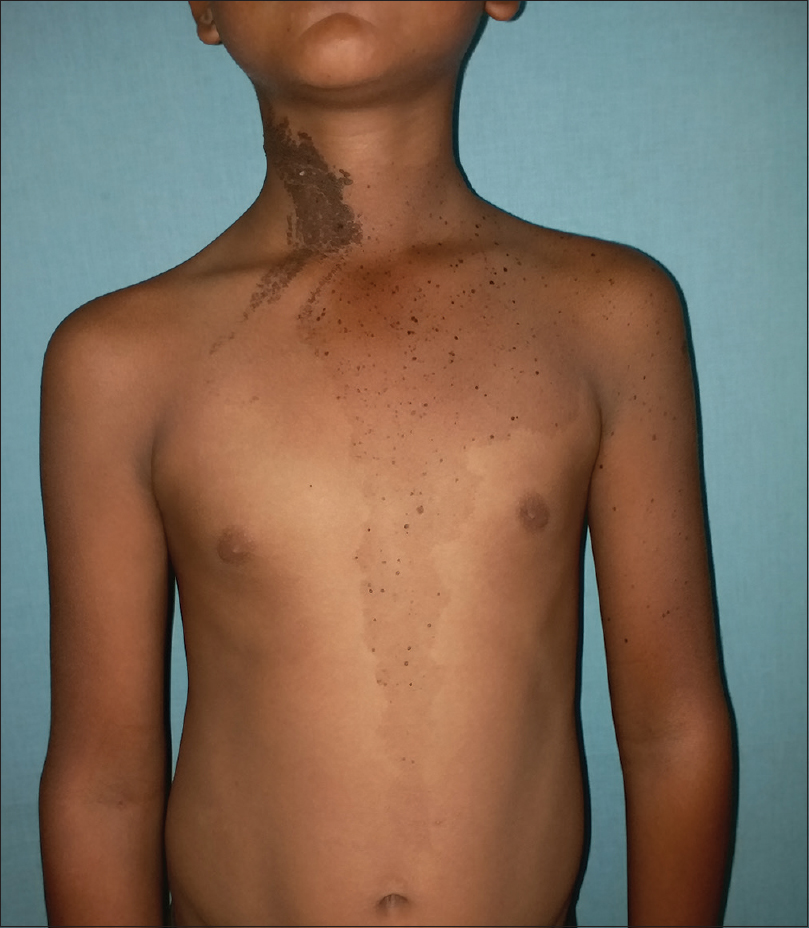 |
| Figure 3: Speckled nevus on the left side of chest wall and upper abdomen |
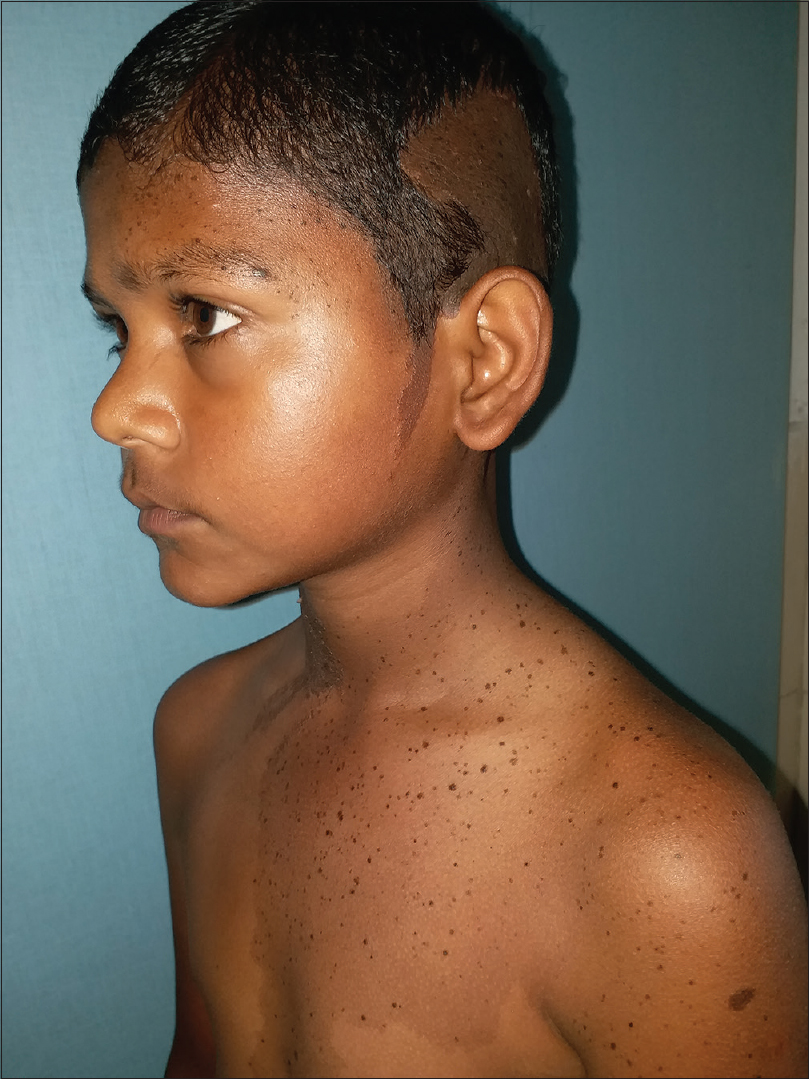 |
| Figure 4: Speckled nevus with multiple small dark brown macules and papules overlying it. Verrucous plaque on left temporal area along with alopecia |
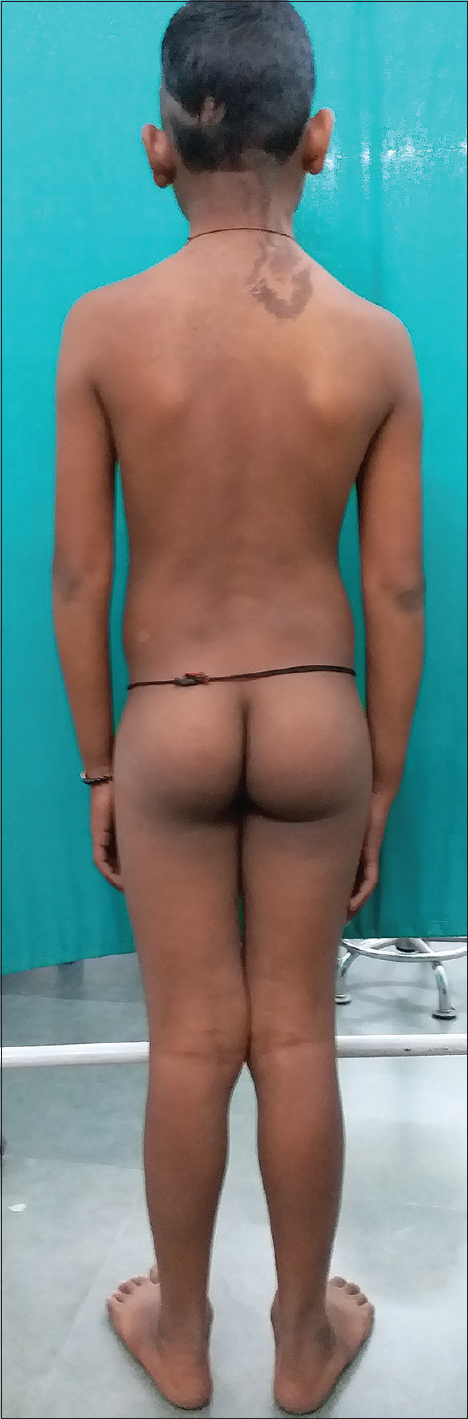 |
| Figure 5: Limb length discrepancy leading to scoliosis |
Skin biopsy of lesions from the neck and scalp showed papillomatosis, hyperkeratosis, orthokeratosis and elongation of rete ridges. Underlying dermis showed melanin incontinence and sparse mononuclear cell infiltrate with an increased number of mature sebaceous glands, confirming the diagnosis of sebaceous nevus [Figure - 6] and [Figure - 7].
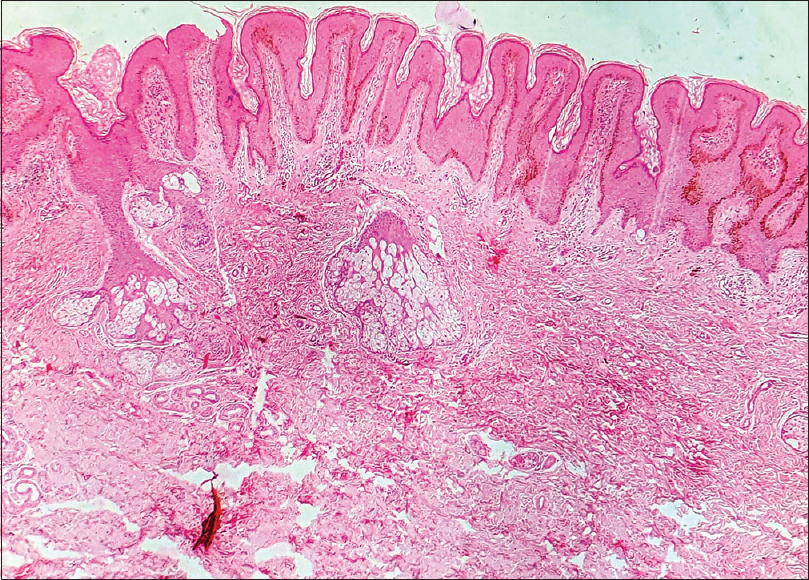 |
| Figure 6: Low power view of nevus sebaceous showing hyperkeratosis, papillomatosis and elongation of rete ridges (H and E, 100×) |
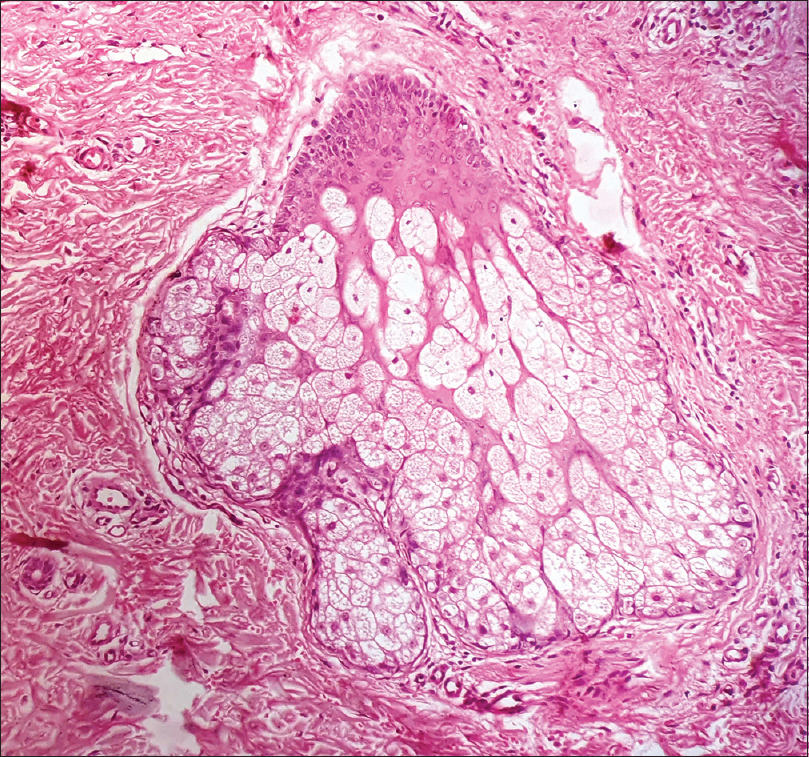 |
| Figure 7: High power view of nevus sebaceous showing mature sebaceous glands (H and E, 400×) |
Serum levels of vitamin D3 and ionized calcium were normal. Ultrasonography of the pelvis revealed a significantly small right kidney (5.22 cm) and a normal left kidney (10.24 cm) which was confirmed by computed tomography scan [Figure - 8]. Hematological and biochemical investigations were normal, except for an elevated alkaline phosphatase level. Ophthalmological, otological and neurological investigations were done to rule out other described extracutaneous associations and no abnormality was detected.
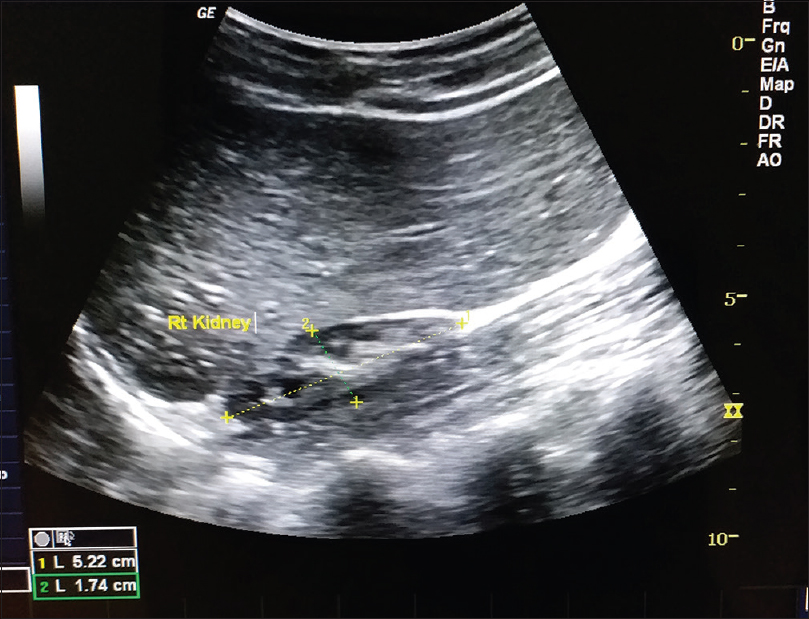 |
| Figure 8: Ultrasonography revealing hypoplastic right kidney |
The patient did not want any treatment for the skin condition. We advised a yearly follow-up to look for the development of malignancy, and renal function tests and ultrasonography of kidneys were also repeated during this visit.
The epidermal nevus syndromes represent a group of distinct disorders that can be recognized and distinguished by the type of associated epidermal nevus.[3],[4] [Table - 1] summarizes the most relevant features of well-defined epidermal nevus syndromes.

Approximately 35 cases of phakomatosis pigmentokeratotica have been reported in the literature. Phakomatosis pigmentokeratotica is characterized by the coexistence of a sebaceous nevus and a speckled lentiginous nevus. In phakomatosis pigmentokeratotica, the sebaceous nevus is preferentially localized in the cranial parts of the body, whereas the keratinocytic epidermal nevus is located commonly on the trunk and extremities.[5] Furthermore, some neurological, skeletal or other extracutaneous abnormalities may be associated.
Until recently, the occurrence of two different nevus types in a single patient was explained by the concept of twin spotting, also referred to as didymosis, as proposed by Happle. As per this theory, the two nevi would have originated from an early event of postzygotic recombination, resulting in a loss of heterozygosity and thus presenting in a homozygous fashion at either of the two loci that are situated in different regions, on a pair of homologous chromosomes.[1] However, Groesser et al. presented molecular findings that disproved the twin spot hypothesis. It is now hypothesized that a single heterozygous activating Harvey rat sarcoma mutation, acting dominantly, accounts for the two distinct nevi.[6] He proposed that in isolated Schimmelpenning syndrome, the underlying mosaic Harvey rat sarcoma mutations get lost in the melanocytic progenitor cells. Conversely, in isolated papular nevus spilus syndrome, the mosaic Harvey rat sarcoma mutation is lost in the epithelial progenitor cells.[4] As the two syndromes occur together, often in the form of phakomatosis pigmentokeratotica, this binary disorder may be categorized as a “pseudodidymosis.”[3] In phakomatosis pigmentokeratotica, the mutated progenitor cell still can differentiate into epithelial cells and melanocytes, whereas in Schimmelpenning syndrome, it has lost the latter. The neurological defects of the Schimmelpenning syndrome involve mental deficiency, seizures and hemiparesis, whereas papular nevus spilus syndrome is more likely to be associated with segmental hyperhidrosis, dysesthesia and sensory or motor neuropathy in the area of the speckled lentiginous nevus.
Other reported extracutaneous presentations of phakomatosis pigmentokeratotica include neurologic disorders like hemiatrophy with muscle weakness, dysesthesia, hyperhidrosis; ophthalmologic defects such as sclerotic nevus spread, internal strabismus, ptosis; and skeletal defects such as postural deviation with kyphosis or scoliosis. Facial dysmorphism, conductive hearing loss and hypophosphatemic vitamin D resistant rickets also have been reported. At puberty, lesions may become verrucous with hyperplastic sebaceous glands. Malignant transformation of cutaneous components of phakomatosis pigmentokeratotica, as well as an association with various internal malignancies like urothelial carcinomas and rhabdomyosarcoma, have also been reported.[7]
In our case, in addition to scoliosis, the cutaneous signs of phakomatosis pigmentokeratotica were associated with unilateral renal hypoplasia. Right renal agenesis has been reported in a case of phakomatosis pigmentovascularis type IIb; however, this is the first case of phakomatosis pigmentokeratotica being reported in association with unilateral renal hypoplasia.[8] Keeping in mind the possibility of malignant transformation of epidermal nevi, the patient is on follow up for the same. It is also prudent to watch out for other anomalies, even in clinically asymptomatic individuals, with a combination of the characteristic nevi of phakomatosis pigmentokeratotica.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form, the legal guardian has given his consent for images and other clinical information to be reported in the journal. The guardian understands that names and initials will not be published and due efforts will be made to conceal the identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
| 1. |
Happle R, Hoffmann R, Restano L, Caputo R, Tadini G. Phakomatosis pigmentokeratotica: A melanocytic-epidermal twin nevus syndrome. Am J Med Genet 1996;65:363-5.
[Google Scholar]
|
| 2. |
Tadini G, Restano L, Gonzáles-Pérez R, Gonzáles-Enseñat A, Vincente-Villa MA, Cambiaghi S, et al. Phakomatosis pigmentokeratotica: Report of new cases and further delineation of the syndrome. Arch Dermatol 1998;134:333-7.
[Google Scholar]
|
| 3. |
Sugarman JL. Epidermal nevus syndromes. Semin Cutan Med Surg 2007;26:221-30.
[Google Scholar]
|
| 4. |
Happle R. The group of epidermal nevus syndromes Part I. Well defined phenotypes. J Am Acad Dermatol 2010;63:1-22.
[Google Scholar]
|
| 5. |
Groesser L, Herschberger E, Sagrera A, Shwayder T, Flux K, Ehmann L, et al. Phakomatosis pigmentokeratotica is caused by a postzygotic HRAS mutation in a multipotent progenitor cell. J Invest Dermatol 2013;133:1998-2003.
[Google Scholar]
|
| 6. |
Groesser L, Herschberger E, Ruetten A, Ruivenkamp C, Lopriore E, Zutt M, et al. Postzygotic HRAS and KRAS mutations cause nevus sebaceous and Schimmelpenning syndrome. Nat Genet 2012;44:783-7.
[Google Scholar]
|
| 7. |
Om A, Cathey SS, Gathings RM, Hudspeth M, Lee JA, Marzolf S, et al. Phakomatosis pigmentokeratotica: A mosaic RASopathy with Malignant Potential. Pediatr Dermatol 2017;34:352-5.
[Google Scholar]
|
| 8. |
Huang C, Lee P. Phakomatosis pigmentovascularis IIb with renal anomaly. Clin Exp Dermatol 2000;25:51-4.
[Google Scholar]
|
Fulltext Views
4,300
PDF downloads
2,242





![[Figure - 1]](#fig_ijdvl_2020_86_5_545_291439_f1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2020_86_5_545_291439_f2.jpg){kind=link}
![[Figure - 3]](#fig_ijdvl_2020_86_5_545_291439_f3.jpg){kind=link}
![[Figure - 4]](#fig_ijdvl_2020_86_5_545_291439_f4.jpg){kind=link}
![[Figure - 5]](#fig_ijdvl_2020_86_5_545_291439_f5.jpg){kind=link}
![[Figure - 6]](#fig_ijdvl_2020_86_5_545_291439_f6.jpg){kind=link}
![[Figure - 7]](#fig_ijdvl_2020_86_5_545_291439_f7.jpg){kind=link}
![[Figure - 8]](#fig_ijdvl_2020_86_5_545_291439_f8.jpg){kind=link}
![[Table - 1]](#tbl_ijdvl_2020_86_5_545_291439_t9.jpg){kind=link}